© Borgis - Nowa Pediatria 1/2003, s. 76-80
Henryk W. Witas, Jerzy Bodalski
Podstawy immunogenetyki w chorobach autoimmunologicznych u dzieci
Immunogenetic background of autoimmunologic diseases in children
z Pracowni Biologii Molekularnej, Kliniki Chorób Dzieci, Instytutu Pediatrii, Uniwersytetu Medycznego w Łodzi
Kierownik Pracownii: dr hab. n. med. Henryk W. Witas
Streszczenie
The term autoimmunity encompasses a variety of polygenic diseases ranging from organ specific disorders such as diabetes mellitus, multiple sclerosis or celiac disease to systemic ones such as lupus erythematosus. Genetic predisposition as well as many environmental factors may contribute to pathogenesis of autoimmune disorders. It seems that the most intriguing feature recently studied, which probably is the main pathology target is lymphocyte activation i.e. primary and secondary signal both generated to get started proliferation of specific T cell clones. A number of molecules involved in T cell activation is known, and others are still being discovered as the search for candidate genes, which form predisposing genotype is continued. This object of direct or indirect studies could show itself as a means of possible therapeutic intervention through the direct modification of genes or protein products of their expression.
Słowa kluczowe: autoimmune disease,
limfocyte activation,
HLA,
TCR,
CD28,
CTLA-4,
ICOS,
PD-1,
TNF,
OX40,
costimulation.

Wprowadzenie
Napadowa, zimna hemoglobinuria, którą po raz pierwszy opisali Donath i Landsteiner w 1904 roku (1) jest pierwszym śladem, który rozpoczął żmudny i długotrwały, jeszcze nie zakończony proces wyjaśniania mechanizmów prowadzących do ujawnienia się chorób autoimmunologicznych. Są one obserwowane stosunkowo rzadko. Występują u ok. 4-7% populacji, według różnych autorów, przy czym częściej u przedstawicieli płci żeńskiej oraz ludzi starszych (2, 3). Do nielicznych wyjątków obserwowanych nieco częściej należą m. in.: cukrzyca insulinozależna, autoimmunologiczne zapalenie tarczycy, reumatoidalne zapalenie stawów. Choroby o podłożu autiommunologicznym stanowią grupę o stosunkowo słabo poznanych mechanizmach powstawania oraz rozwoju bezobjawowego i objawowego. Szereg autorów próbuje definiować je jako zespoły kliniczne wywołane aktywacją limfocytów T lub/i B bez identyfikowalnej przyczyny w postaci infekcji. Z punktu widzenia genetyki dziedziczenie choroby autoimmunologicznej nie jest związane z pojedynczą cechą, która warunkuje jej postać kliniczną, ale dotyczy określonego układu cech, stanowiącego o predyspozycji organizmu. Zadaniem niniejszej publikacji jest przedstawienie udziału czynników biorących udział w przekazywaniu pierwotnego i wtórnego sygnału oraz kodujących je alleli formujących genotyp, który predysponuje do ekspresji patologicznego fenotypu.
AKTYWACJA LIMFOCYTÓW T
Antygeny zarówno obce jak i własne są rozpoznawane po uprzednim ich przetworzeniu w nielicznych tylko komórkach (makrofagi, limfocyty B i komórki dendrytyczne) i następującej potem prezentacji limfocytom CD4+ za pośrednictwem związanych z błoną komórkową hetrodimerycznych białek kodowanych przez geny głównego kompleksu zgodności tkankowej (MHC, HLA). Oprócz prezentowania antygenów komórkom efektorowym układu immunologicznego, cząsteczki HLA spełniają swoją biologiczną funkcję również na powierzchni komórek zrębu, różnicując w procesie delecji klonalnej tymocyty, podczas ich wędrówki pomiędzy korą i rdzeniem grasicy. Stanowią zatem jeden z elementów fizjologicznego systemu kontroli ilości autoreaktywnych komórek T docierających na obwód. Ograniczenie sygnału tylko do oddziaływania typu HLA/antygen/ TCR (sygnał pierwotny) powoduje m.in. to że komórki T nie wydzielają IL-2 i IFNγ osiągając w efekcie stan anergii i nie odpowiadają na bodźce różnicujące. Warunkiem powstania pełnego sygnału do proliferacji komórek T jest nie tylko odpowiednia ilość antygenu uruchamiającego układ HLA-TCR, ale także powstanie poza nim tzw. dodatkowego sygnału do aktywacji (wtórnego, kostymulującego). Istnieje co najmniej kilka par cząsteczek typu ligand-receptor, które biorą udział w jego przekazywaniu. Układ B7 i CD28/CTLA4/ICOS jest najlepiej spośród nich poznanym. Poszczególne składniki sygnału dodatkowego transmitowanego w ten sposób wzmagają, o kilka rzędów wielkości, produkcję różnych limfokin, w tym nie tylko interleukiny 2 (IL-2) odpowiedzialnej za proliferację limfocytów T, ale także GM-CSF, TNF-α, IFN-γ czy IL10 biorącej udział w różnicowaniu komórek B. Proces jest kontrolowany dwojako – zarówno poprzez stymulację transkrypcji właściwych rodzajów mRNA jak i ich stabilizację. Co więcej, ten sam sygnał indukuje ekspresję anty-apoptycznego białka bcl-xL, którego funkcja biologiczna sprowadza się do utrzymania przeżywalności komórek. W procesie aktywacji limfocytów T udział biorą także liczne cytokiny wydzielane przez komórkę prezentującą antygen, w tym IL-1 i IFNα. Pierwsza spośród nich wiąże się z receptorem typu I obecnym na limfocycie T i jest kolejnym białkiem zaangażowanym w aktywację komórki.
SYGNAŁ PIERWOTNY
Regionem, w którym występują najdawniej poznane czynniki genetyczne związane z występowaniem chorób autoagresyjnych jest główny kompleks zgodności tkankowej, zlokalizowany na chromosomie 6.
Antygeny prezentowane limfocytom CD4+ są przyłączane w zagłębieniu, które tworzą zewnętrzne domeny błonowe łańcuchów α i β. Ich wiązanie jest realizowane częściowo poprzez wiązania wodorowe wytwarzane pomiędzy konserwatywnymi aminokwasami tworzącymi zagłębienie i polipeptydem (4, 5). Oddziaływanie tzw. kotwiczących reszt aminokwasowych prezentowanego peptydu z kieszonkami (P1, P4, P6, P7, P9) zbudowanymi z polimorficznych domen wzmacnia lub osłabia wiązanie. Aminokwasy, które są zaangażowane w takie wiązanie mogą wytwarzać wiązania wodorowe pomiędzy azotem grupy amidowej i atomem tlenu grupy karboksylowej prezentowanego peptydu (6).
Powszechnie uważa się, że zależność pomiędzy zawartością poszczególnych kodonów np. HLA DQA1, HLA DQB1, HLA DRB1, a rodzajem odpowiedzi immunologicznej realizowana może być poprzez rodzaj kodowanych aminokwasów, zaangażowanych w tworzenie struktury trzeciorzędowej domen białkowych dimerów prezentujących antygen limfocytom T. Według hipotezy Nepoma (7) zmiana oddziaływań w obrębie kieszonek przyłączających i prezentującej antygen jest odpowiedzialna za siłę jego wiązania (różnice sięgające kilku rzędów wielkości) i może pretendować do roli jednego z mechanizmów kontrolujących eliminację autoreaktywnych tymocytów w grasicy.
SYGNAŁ DODATKOWY (wtórny, kostymulacyjny)
Oddziaływanie typu HLA-antygen-TCR tj. sygnał pierwotny, wymaga wsparcia w postaci współoddziaływania szeregu cząsteczek obecnych na obydwóch rodzajach komórek tj. prezentujących antygen i limfocytach T. Ich aktywność, prezentacja na powierzchni lub uwalnianie na zewnątrz następuje jako bezpośredni efekt pierwotnego kontaktu i jest konieczna co najmniej z trzech powodów: niskiego powinowactwa receptora TCR do związanego z HLA antygenu, utrudnionego oddziaływania pomiędzy elementami kompleksu ze względu na niewielkie rozmiary cząsteczek TCR i HLA oraz ze względu na niewielką liczbę niezmiennych antygenowo rozpoznających kompleksów na komórkach prezentujących (APC). Efektem oddziaływania cząsteczek kostymulujących (białka sygnału dodatkowego), otoczonych wieńcem białek adhezyjnych z rodziny integryn, określanym mianem synapsy immunologicznej, jest utrwalenie kontaktu z receptorem TCR i umożliwienie transmisji sygnału (8).
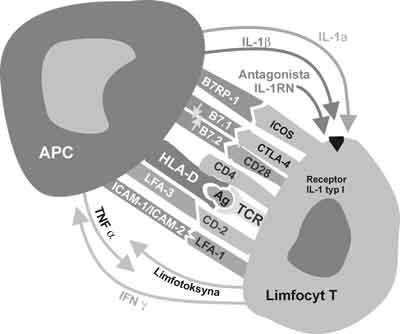
Spośród wielu poznanych dotychczas cząsteczek warunkujących transmisję dodatkowego sygnału do aktywacji limfocytów T omówione poniżej zostaną: CD28, CTLA-4, ICOS, PD-1, TNF, OX40 (ryc. 1).
Ryc. 1. Niektóre białka pośredniczące podczas przekazywania sygnałów – pierwotnego (HLA-Ag-TCR) i dodatkowego, niezbędnych do aktywacji limfocytów T.
CD28, CTLA-4 (ang. cytotoxic T lymphocyte antigen)
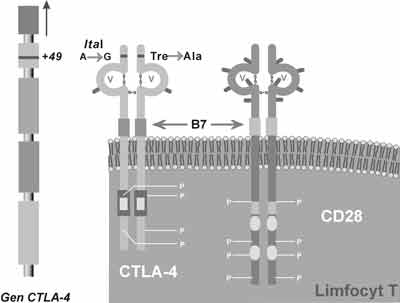
Para cząsteczek typu ligand-receptor CD28-CTLA-4 (CD152) należy do grupy najlepiej poznanych cząsteczek biorących udział w przekazywaniu sygnału pomiędzy immunokompetentnymi komórkami podczas odpowiedzi immunologicznej (9). Należą one do nadrodziny cząsteczek immunoglobulinopodobnych przy czym CD28 obecna jest stale na powierzchni komórek T, a CTLA-4 transportowana jest z cytoplazmy wyłącznie podczas aktywacji komórki (ryc. 2). Kodujące je geny sąsiadują ze sobą na 2 chromosomie (2q33-34), a bliskie ich położenie i uderzające podobieństwo struktury wskazuje na wspólne pochodzenie ewolucyjne. CD28 jest znacznie słabiej wiążącym receptorem (20-100x) niż CTLA-4, chociaż w przeciwieństwie do tego drugiego, obecnym stale i w znacznie większej ilości, na spoczynkowych komórkach T (10).
Ryc. 2. Lokalizacja i struktura białek CTLA-4 i CD28. Zaznaczono znane polimorficzne miejsce w białku CTLA-4.
ICOS (ang. inducible costimulator)
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Donath J., Landsteiner K.: Über paroxymale Hämoglobinurie. 1904, Munch. med. Wochenschr. 51, 1590-1593. 2. Sinha A.A. et al.: Autoimmune diseases: the failure of self tolerance. Science 1990, 248:1380-1388. 3. Jakobson D.L. et al.: Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997, 84:223-143. 4. Madden D.R.: The three-dimensional structure of peptide-MHC complexes. Ann. Rev. Immunol. 1995, 13:587-622. 5. Brodsky F.M. et al.: Antigen processing and presentation. Tissue Antigens 1996, 47:464-471. 6. Hammer J. et al.: HLA class II binding specificity and autoimmunity. Adv. Immunol. 1997, 61: 67-100. 7. Nepom G.T. et al.: Recognition of altered self major histocompatibility complex molecules modulated by specific peptide interactions. Eur. J. Immunol. 1996, 26:949-952. 8. Gracoui A. et al.: The immunological synapse: a molecular machine controling T cel activation. Science, 1999, 285:221-227. 9. Witas H.W. i wsp.: Dodatkowy sygnał w procesie aktywacji limfocytów T. Post. Biol. Kom. 1998, 25:111-123. 10. Racke M.K., Stuart R.W.: Targeting T cell costimulation in autoimmune disease. 2002, 6:275-289. 11. Coyle A.J. et al.: The CD28-related molecule ICOS is reqired for effective T cell-dependent immune responses. Immunity 2000, 13:95-105. 12. Mages H.W. et al.: Molecular cloning and characterization of murine ICOS and identification of B7h as ICOS ligand. Eur. J. Immunol. 2000, 30:1040-1047. 13. McAdam A.J. et al.: Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J. Immunol. 2000, 165: 5035-5040. 14. Hutloff A. Eljaschewitsch B. et al.: ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature, 397:263-266. 15. Shinohara T. et al.: Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics, 1994, 23:704-706. 16. Finger L.R. et al.: The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitore. Gene, 1997, 197:177-187. 17. Freeman G.J. et al.: Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med., 2000, 192:1027-1034. 18. Agata Y. et al.: Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol., 1996, 8:765-772. 19. Paterson D.J. et al.: Antigens of activated rat T lymphocytes including a molecule of 50 000 Mr detected only on CD positive T blasts. Molecular Immunol., 1987, 24:1281-1289. 20. Vihakar R. et al.: Activation-induced expression of human programmed death-1 gene in T-lymphocytes. Exp. Cell Res., 1997, 232:25-28. 21. Latchman Y. et al.: PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature Immunology 2001, 2:261-268. 22. Won B., Choi Y.: Pathways leading to cell death in T cells. Curr. Opin. Immun. 1997, 9:358-364. 23. Thompson C.B.: Apoptosis in the pathogenesis and treatment of the disease. Science 1995, 267:1456-1462. 24. Messer G. et al.: Polymorphic structure of the tumor necrosis factor (TNF) locus: An NcoI polymorphism in the first intron of the human TNFb gene correlates with variant amino acid in position 26 and a reduced level of TNFb producton. J. Exp. Med.
1991, 173:209-219. 25. Wilson A.G. et al.: Single base polymorphism in the human tumor necrosis factor alfa (TNFa) gene detectable by NcoI restriction of PCR product. Hum. Mol. Genet. 1992, 1:353. 26. Witas H.W. et al.: Does TNF locus contribute to coeliac disease as the independent genetic factor or linked to particular HLA-haplotype? Cent. Eur. J. Immunol. 2000, 25:57-62. 27. Nohara C. et al.: Amelioration of experimental encephalomielitis with anti-OX40 ligand monoklonal antibody: critical role for OX40 ligand in migration, but not development, of pathogenic T cells. J. Immunol., 2001, 166:2108-2115. 28. Rogers P.R. et al.: OX40 promoes Bcl-xL and Bcl-2 expresion and is essential for long-term survival of CD4 cells. Immunity, 2001, 15:445-55. 29. Gramaglia I. et al.: The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000, 165:3043-3050. 30. Nepom G.T., Erlich H.: MHC class-II molecules and autoimmunity. Annu Rev. Immunol. 1991, 9:493-525. 31. Kuchroo V.K. et al.: B7.1 and B7.2 costimulatory molecules differentially activate the Th1/Th2 developmental pathways: application to autoimune disease therapy. Cell 1995, 80:707-718. 32. Harper K. et al.: CTLA-4 and CD28 activated lymphocyte molecules are closely related i both mouse and human as to sequence, message expression, gene structure, and chromosomal location. J. Immunol. 1991, 147:1037-1044. 33. Donner H. et al.: CTLA-4 alanine-17 confers genetic susceptibility to Grave´s disease and to type 1 diabetes mellitus. J. Clin. Endocrinol. Metab. 1997, 82:143-146. 34. Huang H.S. et al.: Insulin-dependent diabetes mellitus (IDDM) is asociated with CTLA-4 polymorphism in multiple ethnic groups. Hum. Mol. Genet. 1997, 6:1275-1282. 35. Witas H.W. et al.: The CTLA-4 gene may contribute to coeliac disease. 1998 Biomed. Lett., 57:171-181. 36. Oaks. K. et al.: A naive soluble form of CTLA-4. 2000, 201:144-153.
