© Borgis - Postępy Nauk Medycznych 2-3/2002, s. 117-122
Franciszek Kokot, Rafał Ficek
Rola układu reninowo-angiotensynowo-aldosteronowego (RAA) w patogenezie nadciśnienia tętniczego
Role of the renin-angiotensjon-aldosterone system in the pathogenesis of arterial hypertension
Katedra i Klinika Nefrologii, Endokrynologii i Chorób Przemiany Materii Śląskiej Akademii Medycznej w Katowicach
Kierownik Katedry: prof. dr hab. med. Andrzej Więcek
Streszczenie
Systemowy (krążący we krwi) i lokalne układy reninowo-angiotensynowo-aldosteronowe (RAA) są ważnymi ogniwami regulacji ciśnienia tętniczego w warunkach fizjologicznych. Układy te wykazują istotny wpływ na dwie główne determinanty ciśnienia tętniczego tj. opór naczyń krwionośnych i wolemię. Układy te odgrywają istotną rolę w patogenezie nie tylko wtórnych postaci nadciśnienia tętniczego ale również nadciśnienia samoistnego. Za taką rolą przemawiają wyniki badań doświadczalnych, molekularnych jak i klinicznych. Uzależnienie terapii przeciwnadciśnieniowej blokerami układu RAA tylko od profilu reninowego osocza jest patofizjologicznie nieuzasadnione.
Summary
Both the systemic (circulating) and local renin-angiotensin-aldosterone systems are essential factors participating in the regulation of blood pressure under physiological conditions. These systems are markedly influencing the two main determinants of blood pressure – volaemia and vascular resistance. They are involved not only in the pathogenesis of secondary but also of essential arterial hypertension. The pathogenic role of the RAA in arterial hypertension is confirmed by experimental, molecular and clinical studies. Dependence of usage of RAA blockers in hypertensive patients only on the plasma renin profile is pathophysiologically unjustified.

Historia układu reninowo-angiotensynowo-aldosteronowego (RAA) liczy nieco więcej niż 100 lat. Milowymi etapami w jej rozwoju były następujące odkrycia:
– wykazanie w 1898 r. przez Tigerstedta i Bergmana hipertensyjnego działania wyciągów nerkowych (zawierających reninę),
– stwierdzenie przez Goldblatta w 1934 r., że niedokrwienie nerki może być przyczyną nadciśnienia tętniczego,
– udowodnienie przez E. Brauna-Menendeza i Page´a (1939 r.), że nie renina a angiotensyna jest przyczyną nadciśnienia tętniczego u zwierząt z niedokrwioną nerką,
– wykrycie aldosteronu przez Simpsona, Tait i Wetsteina (1953 r.),
– poznanie struktury angiotensyny I i II przez Skeggsa i wsp. w 1954 r.,
– identyfikacja przez Davisa, Genest, Laragh i innych występowania systemowego układu RAA (1960-1961 r.),
– odkrycie saralazyny, pierwszego peptydowego antagonisty angiotensyny II (1971 r.),
– synteza pierwszego inhibitora konwertazy angiotensyny I – kaptoprylu (1977 r.),
– poznanie struktury receptorów angiotensyny II oraz ich blokerów (wczesne lata osiemdziesiąte).
Celem niniejszej pracy jest zwięzłe podsumowanie aktualnego stanu wiedzy dotyczącego roli układu RAA w patogenezie nadciśnienia tętniczego. W pracy uwzględniono głównie piśmiennictwo ostatnich 5-7 lat, w tym szczególnie prace przeglądowe. Wcześniejsze piśmiennictwo dotyczące tego zagadnienia znajdzie zainteresowany Czytelnik w pracach 39, 40, 41, 46, 47, 48.
GENERACJA I AKTYWNOŚĆ POSZCZEGÓLNYCH OGNIW UKŁADU RENINOWO-ANGIOTENSYNOWEGO (RA)
Jak widać na rycinie 1 wyróżnia się przynajmniej 5 postaci angiotensyn powstających z jednego substratu, tj. angiotensynogenu. Angiotensyny te różnią się liczbą aminokwasów, rodzajem receptora oraz aktywnością biologiczną. W warunkach fizjologicznych głównym enzymem zapoczątkowującym powstawanie angiotensyn jest renina będąca enzymem proteolitycznym, katalizującym powstawanie dekapeptydu – angiotensyny-1-10 (ang-1-10), określana również jako angiotensyna I. Z kolei angiotensyna I jest substratem dla względnie nieswoistej proteazy – konwertazy, katalizującej przekształcenie angiotensyny I w oktapeptyd – angiotensynę II (ang-1-8). Konwertaza angiotensyny I katalizuje również rozkład innych biologicznych aktywnych polipeptydów takich jak bradykinina. Jak widać na rycinie 1 angiotensyna II może również powstać bezpośrednio z angiotensynogenu (pod wpływem takich enzymów jak tonina, elastazam, katepsyna G i enzym generujący angiotensynę II wrażliwy na chymostatynę – CAGE) lub też z angiotensyny I (pod wpływem takich enzymów jak chymaza, tonina lub katepsyna G). Z kolei z angiotensyny II może powstać peptyd siedmioaminokwasowy – angiotensyna-2-8 (tzw. angiotensyna III) lub sześcioaminokwasowy – angiotensyna-3-8 (zwana również angiotensyną IV). Piątą angiotensyną jest polipeptyd złożony z 7 N-końcowych aminokwasów angiotensyny I i II, określano jako angiotensyna-1-7.
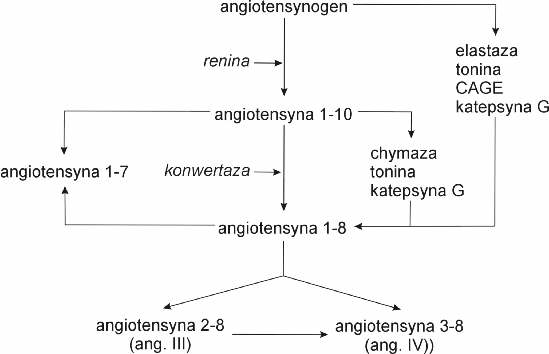
Ryc. 1. Szlaki syntezy poszczególnych angiotensyn. CAGE – enzym generujący angiotensynę II wrażliwy na chymostatynę.
Angiotensyna-1-10 nie wydaje się posiadać znaczenia fizjologicznego, bowiem ulega szybkiemu przekształceniu przez wszędobylską konwertazę.
Angiotensyna II jest oktapeptydem o wielokierunkowym działaniu. Stymuluje skurcz miocytów naczyniowych i przebudowę naczyń krwionośnych, uwalnianie tlenku azotu, prostacykliny, endoteliny i tromboksanów przez śródbłonek naczyniowy, proces aterogenezy naczyń, przerost kardiomiocytów, uczucie pragnienia, sekrecję wazopresyny i aldosteronu oraz aktywność fibroblastów w nerkach i mięśniu sercowym (prowadząc do włóknienia tych narządów). Ponadto hormon ten stymuluje układ sympatyczny, pobudza ekspresję genu kodującego inhibitor aktywatora plazminogenu (działając antyfibrynolitycznie), natomiast hamuje procesy apoptyczne przez co sprzyja proliferacji różnych komórek m.in. komórek immunologicznie kompetentnych (ryc. 2). Hormon ten nie tylko wpływa na hemodynamikę nerek (spadek ukrwienia nerek), ale również na wchłanianie zwrotne sodu przez cewki nerkowe (pobudza resorpcję zwrotną sodu) (29, 42, 60, 64, 69, 88).
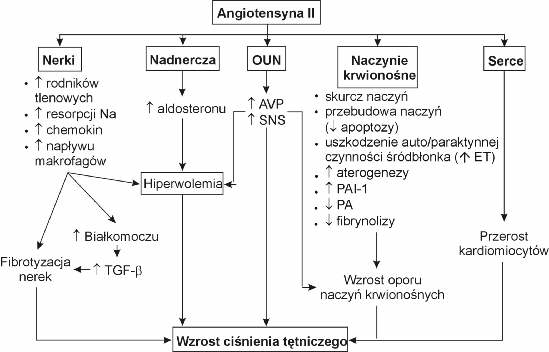
Ryc. 2. Efekty biologiczne angiotensyny II; TGF-b – czynnik wzrostowy transformujący beta; AVP – wazopresyna; SNS – układ sympatyczny; ET – endotelina; PA – aktywator plazminogenu; PAI – inhibitor aktywatora plazminogenu
Receptorami dla angiotensyny II są receptor AT1 i AT2 (3, 7, 12, 13, 87, 94). Stymulacja receptorów AT1 wywołuje ww. zmiany humoralne, hormonalne, sercowo-naczyniowe, nerkowe i nerwowe. Efekty antagonistyczne do wyżej wymienionych wywołuje stymulacja receptorów AT2 (działanie antyproliferacyjne, proapoptotyczne, wazodylatacja, działanie natriuretyczne) (12, 94). Obecność receptorów zarówno AT1 jak i AT2 stwierdzono w sercu, naczyniach krwionośnych, nerkach i ośrodkowym układzie nerwowym (3, 12, 73). Podkreślić należy, że ekspresja receptorów AT2 jest najbardziej zaznaczona w życiu płodowym (36).
Angiotensyna-2-8 wykazuje podobne właściwości co angiotensyna-1-8 (65, 67). Receptor angiotensyny-2-8 różni się od receptora AT1 i A2. Jego struktura nie została jeszcze dokładnie poznana.
Angiotensyna-3-8 określana również jako angiotensyna IV może powstać zarówno z angiotensyny III jak i angiotensyny II. Receptor dla angiotensyny IV wykazuje pewne powinowactwo również do angiotensyny-1-7 (5). Aktywacja receptora AT4 występuje głównie w naczyniach krwionośnych, nerkach i ośrodkowym układzie nerwowym (5).
Angiotensyna-1-7 pobudza syntezę i uwalnianie wazodylatacyjnie działających prostaglandyn, potęguje efekty metaboliczne bradykininy i wzmaga syntezę tlenku azotu (79, 82, 90). Działanie angiotensyny-1-7 jest przeciwstawne do występującego po podaniu angiotensyny II (27, 63). Ponieważ angiotensyna-1-7 ulega rozłożeniu przez enzym konwertujący, po podaniu inhibitorów tego enzymu obserwuje się wzrost jej stężenia we krwi (38, 63, 92). Działanie angiotensyny-1-7 na komórki jest pośredniczone przez receptory inne niż AT2 (26).
Aldosteron jest ważnym ogniwem układu RAA. Odgrywa on istotną rolę w gospodarce wodnoelektrolitowej stymulując resorpcję sodu w nerkach. Ponadto działa tonizująco na naczynia krwionośne oraz dodatnio inotropowo na serce (93). Jego działanie na komórki jest pośredniczone przez receptory cytoplazmatyczne oraz błonowe, będące integralnymi białkami błony komórkowej. Aldosteron nasila procesy włóknienia w sercu i najpewniej również w naczyniach krwionośnych (44).
ROLA UKŁADU RAA W REGULACJI CIŚNIENIA TĘTNICZEGO W WARUNKACH FIZJOLOGICZNYCH
Wyróżnia się dwa rodzaje układu RAA – systemowy, zależny głównie od aktywności aparatu przykłębuszkowego w nerkach oraz tkankowe, obecne prawie we wszystkich narządach (ośrodkowy układ nerwowy, nerki, serce, śródbłonek naczyniowy, tkanka tłuszczowa, nadnercza, łożysko) (35). Aktywność systemowego układu RAA zależna jest od wolemii i ciśnienia perfuzyjnego nerek. Hipoperfuzja nerek spowodowana hipowolemią, spadkiem ciśnienia tętniczego lub niedokrwieniem (zwężenie tętnic nerkowych), zubożenie ustroju w sód oraz aktywacja układu sympatycznego są czynnikami pobudzającymi sekrecję reniny przez aparat przykłębuszkowy. Renina działając na angiotensynogen, katalizuje powstawanie angiotensyny-1-10. Ta ostatnia, ulegając proteolitycznemu działaniu konwertazy, jest źródłem syntezy angiotensyny II. Angiotensyna II działając na nerki (retencja sodu), naczynia krwionośne (skurcz naczyń), serce (dodatnie działanie ino- i batmotropowe), układ sympatyczny (stymulacja), ośrodkowy układ nerwowy (stymulacja sekrecji wazopresyny) i nadnercza (wzrost sekrecji aldosteronu) powoduje nie tylko wzrost wolemii, ale również oporu naczyń krwionośnych. W ten sposób układ RAA stanowi ważne ogniwo regulacji dwóch głównych determinantów ciśnienia tętniczego tj. wolemii i oporu naczyń krwionośnych. Ponadto, systemowy układ RAA stanowi ważny mechanizm czuwający nad homeostazą wodnoelektrolitową (ściśle związaną z regulacją ciśnienia tętniczego), ale również kwasowo-zasadową.
W odróżnieniu od systemowego układu RAA znacznie mniej poznana jest funkcja i regulacja tzw. tkankowych układów RAA. Jak to podano powyżej, występowanie poszczególnych ogniw RAA (angiotensyny, reniny, konwertazy, aldosteronu) w takich narządach jak nerki, nadnercza, serce, naczynia krwionośne, mózg i inne, nie budzi już żadnej wątpliwości. Wymienione ogniwa mogą być syntetyzowane na miejscu lub pochodzić z krążącego we krwi RAA. W oparciu o wyniki wielu badań, głównie doświadczalnych, przyjmuje się, że działanie lokalnych (tkankowych) układów RAA ograniczone jest do komórek wytwarzających poszczególne ogniwa układu RAA (działanie autokrynne) lub komórek sąsiadujących (działanie parakrynne). I tak w OUN angiotensyna II pobudza pragnienie i wydzielanie wazopresyny, w naczyniach krwionośnych uwalnianie związków o działaniu wazopresyjnym (endotelina, aminy katecholowe) jak i naczyniorozkurczowym (bradykinina, tlenek azotu, prostacyklina), w sercu – przerost kardiomiocytów i uwalnianie czynników wzrostowych (powodujących włóknienie mięśnia sercowego), w nerkach – uwalnianie cytokin (TGF-b), chemokin (MCP-1) i czynników wzrostowych (PDGF) (43, 44).
ROLA I FUNKCJA POSZCZEGÓLNYCH OGNIW UKŁADU RAA W ŚWIETLE BADAŃ GENETYCZNYCH
Wiele nowych elementów poznawczych w dziedzinie funkcji i roli układu RAA wniosły badania zwierząt z indukowaną insercją (knock-in) lub delecją (knock-out) genu kodującego określone ogniwo układu RAA.
I tak wykazano u transgenicznych szczurów z mREN2 występowanie złośliwego nadciśnienia tętniczego oraz zwiększenie ekspresji reniny z dużym stężeniem angiotensyny II w takich narządach jak serce, nadnercza i miocyty naczyniowe, pomimo obecności małego stężenia angiotensyny II w surowicy krwi (8, 20, 33, 71). Wyniki tych badań podkreślają znaczącą rolę tkankowych układów RAA w powstawaniu nadciśnienia tętniczego.
Delecja genu kodującego angiotensynogen u zwierząt manifestuje się spadkiem systemowego ciśnienia tętniczego, dużą śmiertelnością pourodzeniową oraz zmianami naczyniowymi nerek u zwierząt dożywających wieku dorosłego (3, 20, 25). Przy nadmiernej ekspresji genu dla angiotensynogenu nie stwierdza się nadciśnienia tętniczego, lecz wzrost ekspresji TGF-b i kolagenu śródmiąższowego indukowanego nefropatią obturacyjną (25).
Myszy pozbawione genu dla ACE1 wykazują małe ciśnienie tętnicze, upośledzenie zagęszczania moczu, zanik rdzenia i brodawek nerkowych, zgrubienie naczyń nerkowych i poszerzenie kielichów nerkowych (15, 20). Zwierzęta pozbawione genu dla ACE2 charakteryzują się normalną budową nerek, chociaż stwierdza się pogrubienie ścian naczyń nerkowych (15). Zarówno delecji genu ACE1 jak i ACE2 towarzyszy wzrost kaliemii i spadek wartości hematokrytowej (15).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Adam A., Raij L.: J. Nephrol. 2000, 12, 211.
2. Adamczak M. i wsp.: J. Hum. Hypertens. 2000, 14, 503.
3. Allen A.M. et al.: Am. J. Hypertens. 2000, 13, 31S.
4. Andersen S. et al.: Kidney Int. 2000, 57, 601.
5. Ardaillou R., Chansel D.: In Renin-Angiotensin. A Centenary symposium of the discovery of the renin angiotensin system. Ulfendahl H.R., Aurell M. (Eds.): Portland Press, London, 1998, 47.
6. Arima S., Ito S.: Nephrol. Dial. Transplant. 2000, 15, 448.
7. Audoly L.P. et al.: TEM 2000, 11, 263.
8. Baltatu O. et al.: W: Renin-Angiotensin. A centenary Symposium of the discovery of the renin-angiotensin system. (red.) Ulfendahl H.R., Aurell M., Portland Press, London, 1998, 105.
9. Berger S. et al.: Kidney Int. 2000, 57, 1295.
10. Bonny O., Hummler E.: Kidney Int. 2000, 57, 1313.
11. Buemi M. et al.: Am. J. Hypertens. 2000, 12, 450.
12. Burnier M., Brunner H.R.: Lancet 2000, 355, 637.
13. Cervenka L. et al.: Kidney Int. 1999, 56, 1855.
14. Chan J.C.N. et al.: Kidney Int. 2000, 57, 590.
15. Cole J., Bernstein K.E.: JRAAS 2000, 1, 137.
16. Conlin P.R. et al.: Am. J. Hypertens. 2000, 13, 418.
17. Corvol P., Jeunemaitre X.: W: Renin-Angiotensin. A Centenary Symposium of the discovery of the renin-angiotensin system. (red.) Ulfendahl H.R., Aurell M., Portland Press, London, 1998, 237.
18. Curtis J.J. et al.: J. Am. Soc. Nephrol. 2000, 11, 2404.
19. Cusi D., Bianchi G.: Kidney Int. 1998, 54, 328.
20. Cvetkovic B., Sigmund C.D.: Kidney Int. 2000, 57, 863.
21. Delcayre C. et al.: Kidney Int. 2000, 57, 1346.
22. Delyani J.A.: Kidney Inter. 2000, 57, 1408.
23. Dzida G. i wsp.: Pol. Arch. Med. Wewn. 1999, 102, 1033.
24. Farman N., Bocchi B.: Kidney Int. 2000, 57, 1364.
25. Fern R. et al.: J. Clin. Invest. 1999, 103, 39.
26. Ferrario C. et al.: J. Am. Soc. Nephrol. 1998, 9, 1716.
27. Ferrario C.M.: J. Nephrol. 1998, 11, 278.
28. Frohlich E.D.: Am. J. Hypertens. 2000, 13, 39.
29. Gibbons G.H.: Am. J. Hypertens. 1998, 11, 177.
30. Gomez R.A.: Kidney Int. 1998, 54, supl. 67, 12.
31. Ha S.K. et al.: Nephrol. Dial. Transplant 2000, 15, 1617.
32. Hatakeyama H. et al.: Kidney Int. 2000, 57, 1352.
33. Hilgers K.F. et al.: Hypertension 1992, 19, 687.
34. Hilgers K.F. et al.: Kidney Int. 2000, 58, 2408.
35. Horiucki M. et al.: In: Renin-Angiotensin. A Centenary Symposium of the discovery of the renin angiotensin system. Ulfendahl H.R., Aurell M. (Eds.): Portland Press, London, 1998, 13.
36. Inagami T.: W: Renin-Angiotensin. A centenary Symposium of the discovery of the renin-angiotensin system. (red.) Ulfendahl H.R., Aurell M., Portland Press, London, 1998, 25.
37. Ittersum F.J. et al.: Nephrol. Dial. Transplant. 2000, 15, 1000.
38. Iyer S.N. et al.: Hypertension 1998, 31 (part 2), 356.
39. Januszewicz A.: Nadciśnienie tętnicze: diagnostyka i leczenie. Rhone-Poulenc, Warszawa 1994.
40. Januszewicz A.: Medipress Kardiologia 1994, 1, 2.
41. Januszewicz W. i wsp.: Nadciśnienie tętnicze. PZWL Warszawa 1993.
42. Klahr S., Morrissey J.J.: Kidney Int. 2000, 57, supl. 75, 7.
43. Kokot F., Ficek R.: Pol. Arch. Med. Wewn. 1999, 101, 289.
44. Kokot F., Ficek R.: Adv. Clin. Exp. Med. 1998, 7, 13.
45. Kokot F., Ficek R.: Macedonian medical review (1997), Suppl. 35, 44.
46. Kokot F., Kokot J.: Postępy Hig. Med. Doświad. 1994, 48, 645.
47. Kokot F., Kokot T.: Post. Nauk Med. 1996, 9, 138.
48. Kokot F.: Kardiologia Polska 1992, 36, 291.
49. Kurokawa K.: Kidney Int. 1998, 54, supl. 67, 71.
50. Lafayette R.A.: Am. J. Kidney Dis. 2000, 35, 166.
51. Laragh J.H., Sealey J.E.: In Renin-Angiotensin. A Centenary Symposium of the discovery of the renin angiotensin system. Ulfendahl H.R., Aurell M. (Eds.) Portland Press, London 1998, 273.
52. Luft F.C.: Current Opinion in Nephrology and Hypertension 2000, 9, 259.
53. Mathiesen E.R. et al.: Brit. Med. J. 1999, 319, 24.
54. Navar L.G. et al.: Am. J. Hypertens. 2000, 13, 45.
55. Oparil S.: Am. J. Hypertens. 2000, 13, 18.
56. Opolski G., Filipiak K.J.: Leki hamujące układ renina-angiotensyna-aldosteron. Urban & Partner, Wrocław, 2000 (wyd. 1).
57. Paran E. et al.: Curr. Opin. Nephrol. Hypertens. 1995, 4, 295.
58. Poch E. et al.: Am. J. Hypertens. 2000, 13, 648.
59. Pontremoli R. et al.: Kidney Int. 2000, 57, 561.
60. Pratt R.E.: J. Am. Soc. Nephrol. 1999, 10, (supl. 12), 120.
61. Preston R.A.: Am. J. Hypertens. 1999, 12, 19.
62. Retting R., Uber A.: Nephrol-Dial-Transplant. 1995, 10 Suppl. 9, 9.
63. Rock A.J. et al.: Hypertension 1999, 34, 296.
64. Rosenberg M.E. et al.: Kidney Int. 1994, 45, 403.
65. Rowe B.P., Dixon B.: Hypertension 2000, 35, 130.
66. Ruilope L.M.: Curr. Opin. Nephrol. Hypertens. 1997, 6, 152.
67. Ruiz-Ortega M. et al.: Kidney Int. 1998, 54, supl. 68, 41.
68. Safian R.D., Textor S.C.: N. Engl. J. Med. 2001, 344, 431.
69. Schnermann J., Briggs J.P.: J. Clin. Invest. 1999, 104, 1007.
70. Schnermann J. et al.: Kidney Int. 1998, 54, supl. 67, 40.
71. Shimokama T. et al.: Virchovs Arch. 1998, 432, 169.
72. Siragy H.M. et al.: J. Clin. Invest. 1999, 104, 181.
73. Smith R. et al.: Emerging Drugs 1998, 3, 81.
74. Smithies O. et al.: Kidney Int. 2000, 58, 2265.
75. Spence J.D.: Am. J. Hypertens. 1992, 12, 1077.
76. Staub O. et al.: Kidney Intern. 2000, 57, 809.
77. Stenvikel P.: Nephrol. Dial. Transplant. 2000, 15, 1115.
78. Stolarczyk D. i wsp.: Postępy Farmakoterapii 2000, 1, 26.
79. Strawn W.B. et al.: Hypertension 1999, 33 (part II), 207.
80. Sugaya T. et al.: J. Biol. Chem. 1995, 270, 18719.
81. Taddei S. et al.: J. Nephrol. 2000, 13, 205.
82. Tallant E.A. et al.: Hypertension 1999, 34 (part 2), 950.
83. Timberlake D.S. et al.: Curr. Opin. Nephrol. Hypertens. 2001, 10, 71.
84. Traynor T. et al.: W: Renin-Angiotensin. A centenary Symposium of the discovery of the renin-angiotensin system. (red.) Ulfendahl H.R., Aurell M. Portland Press, London 1998, 181.
85. Tsusumi Y.: J. Clin. Invest. 1999, 104, 925.
86. Tsutsumi Y. et al.: J. Clin. Invest. 1999, 104, 925.
87. Unger T. et al.: J. Hypertension 1996, 14 supl. 5, 95.
88. Weir M.R., Dzau V.: Am. J. Hypertens. 1999, 12, 205.
89. Whelton P.K. et al.: Curr. Opin. Nephrol. Hypertens. 1997, 6, 177.
90. Widdop R.E. et al.: Hypertension 1999, 34 (part 2), 964.
91. Wiliams G.H.. Fisher N.D.L.: Curr. Opin. Nephrol. And Hypertens. 1997, 6, 199.
92. Yamada K. et al.: Hypertension 1998, 32, 496.
93. Zdrojewicz Z. i wsp.: Nadciśnienie Tętnicze 2000, 4, 209.
94. Żukowska-Szczechowska E. i wsp.: Pol. Arch. Med. Wewn. 1999, 102, 1111.
