© Borgis - Postępy Nauk Medycznych 3/2014, s. 192-196
*Karolina Wejnarska, Elwira Kołodziejczyk, Jarosław Kierkuś, Grzegorz Oracz
Dziedziczne zapalenie trzustki
Hereditary pancreatitis
Klinika Gastroenterologii, Hepatologii i Zaburzeń Odżywiania, Instytut „Pomnik-Centrum Zdrowia Dziecka”, Warszawa
Kierownik Kliniki: prof. dr hab. med. Józef Ryżko
Streszczenie
Dziedziczne zapalenie trzustki jest chorobą rzadką, o autosomalnym dominującym mechanizmie dziedziczenia z około 80% penetracją. Po raz pierwszy zostało opisane w 1952 roku przez Comforta oraz Steinberga, natomiast w 1996 roku zostało powiązane przez Whitcomba i wsp. z mutacją genu trypsynogenu PRSS1. Za najczęściej występujące mutacje uważa się R122H oraz N29I, które stanowią około 90% wykrywanych mutacji. W przypadku Europy Środkowej, w tym Polski, drugą pod względem częstości występowania, po R122H, jest mutacja R122C. Charakterystyczny dla dziedzicznego zapalenia trzustki jest wczesny początek objawów – około 10 roku życia. Przebieg choroby jest zróżnicowany. Z piśmiennictwa wynika, że dziedziczne zapalenie trzustki przez długie lata może przebiegać łagodnie, jednak z dużym prawdopodobieństwem doprowadzi do rozwoju ciężkich powikłań przewlekłego zapalenia trzustki, takich jak niewydolność zewnątrz- i wewnątrzwydzielnicza oraz rak trzustki. Leczenie dziedzicznego zapalenia trzustki nie różni się od leczenia przewlekłego zapalenia trzustki o innej etiologii i polega na leczeniu objawów choroby oraz unikaniu czynników mogących wywołać zaostrzenie. Często niezbędne jest leczenie zabiegowe – endoskopowe i/lub chirurgiczne. Około 50-krotny wzrost ryzyka zachorowania na raka trzustki u pacjentów z dziedzicznym zapaleniem trzustki sprawia, że chorzy ci powinni być objęci czujnym nadzorem onkologicznym i według zaleceń mieć wykonywane regularne, raz do roku, badania – EUS oraz marker CA 19-9 – już od 40 roku życia. Konieczna jest również edukacja pacjentów co do unikania używek, szczególnie palenia papierosów, które, jak wykazały badania, jest czynnikiem najbardziej zwiększającym ryzyko wystąpienia raka trzustki. Rak trzustki istotnie zwiększa śmiertelność u pacjentów z HP.
Summary
Hereditary pancreatitis is a rare autosomal dominant disorder with a penetrance of approximately 80%. HP was first described in 1952 by Comfort and Steinberg, and in 1996 associated with PRSS1 gene mutation by Whitcomb et al. The most frequent mutations of PRSS1 gene are R122H and N29I, which constitute about 90% of the mutations encountered. In Central Europe the second most common mutation after R122H is R122C. Characteristic for hereditary pancreatitis is an early onset of the disease, with median age 10 years. The studies revealed that the clinical course of HP may be mild for many years, but with great probability it leads to severe complications of chronic pancreatitis, such as endocrine and exocrine insufficiency and pancreatic cancer. Treatment of hereditary pancreatitis involves pain management, preventing acute attacks, treatment of maldigestion/malnutrition, diabetes mellitus and local organ complications. Endoscopic and surgical interventions are often needed. Risk of the pancreatic cancer is about 50-fold increased. Therefore, HP patients should strongly avoid environmental risk factors for PC, especially smoking. Pancreatic cancer substantially increases mortality in patients with hereditary pancreatitis. According to the recommendations, HP patients after 40 years of age should have performed EUS and CA 19-9 marker level once a year.

Wstęp
Dziedziczne zapalenie trzustki (ang. hereditary pancreatitis – HP) jest chorobą rzadką, o nieustalonej dokładnie częstości występowania. Dane z piśmiennictwa są zróżnicowane, oceniając częstość tej choroby od 0,3 na 100 000 w populacji francuskiej (1) do 1% pacjentów z przewlekłym zapaleniem trzustki (PZT) w badaniu duńskim (2). Po raz pierwszy zostało ono opisane w 1952 roku przez Comforta oraz Steinberga, którzy zauważyli, że w pewnych rodzinach choroby trzustki występują z większą częstością niż w ogólnej populacji oraz zasugerowali ich genetyczne podłoże (3). Teoria ta została potwierdzona w 1996 roku przez Whitcomba i wsp., którzy odkryli mutację genu trypsynogenu PRSS1 (ang. cationic trypsinogen/serine protease 1) zlokalizowaną na długim ramieniu chromosomu 7, otwierając tym samym rozdział diagnostyki molekularnej w chorobach trzustki, który do dziś nie jest zamknięty (4). Dziedziczne zapalenie trzustki, choć często przebiega łagodnie, to po wielu latach trwania choroby u większości pacjentów prowadzi do poważnych powikłań, takich jak niewydolność zewnątrzwydzielnicza trzustki, cukrzyca czy do najpoważniejszego – raka trzustki, którego ryzyko u chorych z HP jest 50-krotnie wyższe niż ryzyko populacyjne.
Etiologia
Według dzisiejszej wiedzy, dziedziczne zapalenie trzustki jest opisywane jako choroba o autosomalnym dominującym mechanizmie dziedziczenia z około 80% penetracją (5). Jednak piśmiennictwo podaje wartości od 40% w hiszpańskiej pracy de las Heras-Castano i wsp., do ponad 90% w badaniu Reboursa i wsp. (1, 6). Być może nieznane czynniki środowiskowe czy genetyczne mają wpływ na różnorodność penetracji genu w różnych populacjach, jednak mechanizm ich działania nie jest jeszcze poznany.
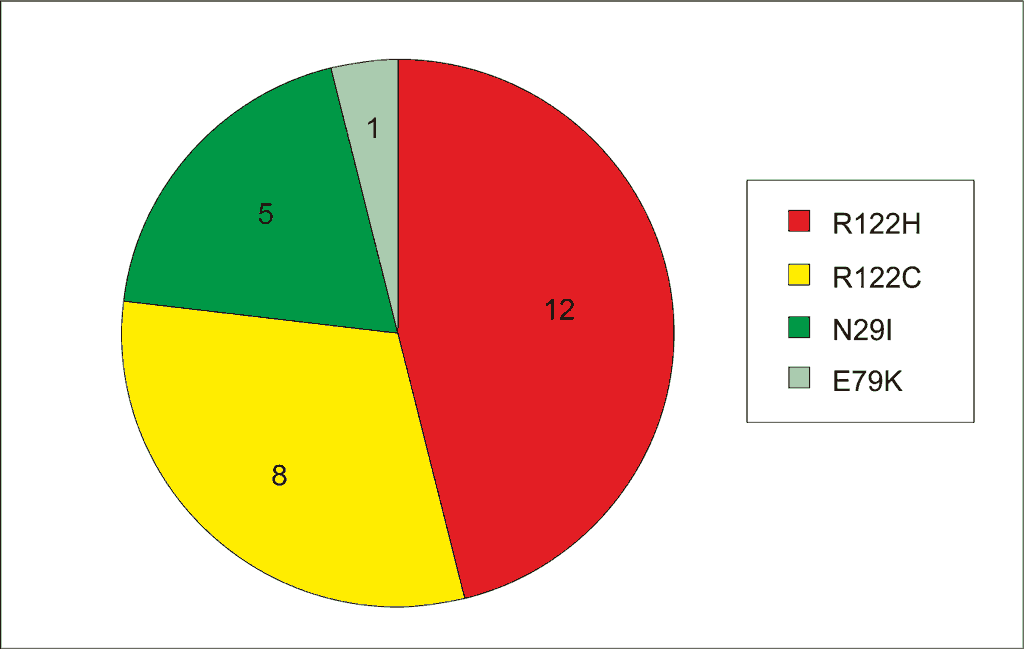
Pierwszą opisaną w 1996 roku mutacją genu PRSS1 była R122H (7), w kolejnych latach sukcesywnie opisywano następne – A16V, D22G, K23R, N29I, N29T, R122C, E79K (8-13). Obecnie za najczęściej występujące uważa się R122H oraz N29I, które według Reboursa i wsp. stanowią około 90% wykrywanych mutacji (1). W przypadku Europy Środkowej, w tym Polski, drugą pod względem częstości mutacją w genie PRSS1, po R122H, jest mutacja R122C. Jest to mutacja dość często obserwowana w Polsce, na terenie Niemiec i w Szwecji (14). Częstość mutacji w genie PRSS1 wśród pacjentów hospitalizowanych w naszej Klinice przedstawia rycina 1.
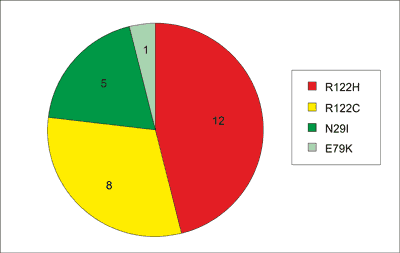
Ryc. 1. Rozkład mutacji w genie PRSS1 u chorych z rodzinnym zapaleniem trzustki hospitalizowanych w CZD.
Mutacja genu kationowego trypsynogenu PRSS1 jest uznana za bezpośredni czynnik sprawczy wystąpienia zapalenia trzustki, w przeciwieństwie do innych mutacji genowych wiązanych z chorobami trzustki – SPINK1 (ang. serine protease inhibitor Kazal type 1), CFTR (ang. cystic fibrosis transmembrane conductance regulator), CTRC (ang. chymotrypsin C) czy CPA1 (ang. carboxypeptidase A1), które są jedynie czynnikami predysponującymi do PZT (15-20).
Mechanizm, w jakim mutacje w genie PRSS1 prowadzą do zapalenia trzustki, był przedmiotem wielu badań. Wydaje się, że kluczowa jest przedwczesna aktywacja kationowego trypsynogenu w przewodach wyprowadzających, co prowadzi do samotrawienia narządu oraz postępującego stanu zapalnego i włóknienia (13, 21). Ustalono również związek mutacji genu PRSS1 ze zmienioną regulacją aktywacji trypsynogenu zależną od chymotrypsyny C (22).
Wpływ czynników środowiskowych na rozwinięcie oraz przebieg dziedzicznego zapalenia trzustki był wielokrotnie badany i jest dotąd nieustalony. Wydaje się, że wbrew wcześniejszym podejrzeniom, używki takie jak papierosy czy alkohol nie rzutują na ciężkość przebiegu dziedzicznego zapalenia trzustki (16). Jednak bezsprzecznie udowodniono, że palenie papierosów u chorych z HP powoduje wzrost ryzyka zachorowania na raka trzustki (23).
Rozpoznanie
Kryteria rozpoznania HP zmieniały się w ciągu ostatnich dekad. Obecnie obowiązujące zostały zdefiniowane przez EUROPAC (The European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer). W przypadku braku genetycznie potwierdzonej mutacji w genie PRSS1, by rozpoznać dziedziczne zapalenie trzustki konieczne jest wystąpienie ostrego nawracającego zapalenia trzustki i/lub przewlekłego zapalenia trzustki o nieznanej etiologii u dwóch krewnych pierwszego stopnia lub trzech (lub więcej) krewnych drugiego stopnia w dwóch lub więcej pokoleniach. U chorych z potwierdzoną mutacją genu PRSS1 oraz obrazem klinicznym przewlekłego zapalenia trzustki rozpoznanie HP jest oczywiste. U pacjentów, którzy nie spełniają podanych kryteriów, a więcej niż jeden członek ich rodziny w tym samym pokoleniu choruje na PZT, rozpoznajemy rodzinne przewlekłe zapalenie trzustki (16). Według zaleceń Polskiego Klubu Trzustkowego chorzy z pozytywnym wywiadem rodzinnym HP powinni mieć wykonane badania genetyczne (24).
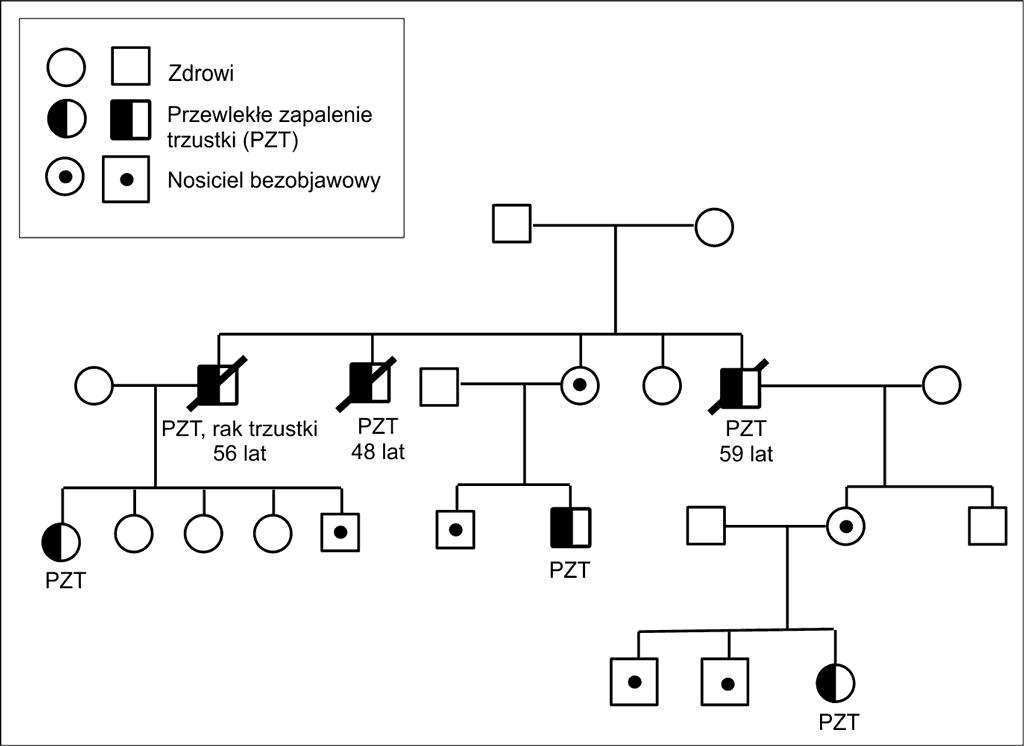
U znacznego odsetka chorych z HP wywiad rodzinny jest bardzo obciążony. Drzewo genealogiczne typowej rodziny z HP przedstawia rycina 2.
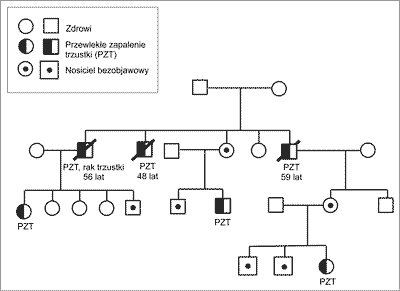
Ryc. 2. Drzewo genealogiczne rodziny z mutacją R122C w genie PRSS1.
Przebieg kliniczny
Przebieg kliniczny dziedzicznego zapalenia trzustki jest zróżnicowany. Zazwyczaj choroba rozpoczyna się jako ostre zapalenie trzustki, następnie poprzez ostre nawracające zapalenie trzustki, które jest uważane za stadium przejściowe choroby, rozwija się do przewlekłego zapalenia trzustki z typowymi, utrwalonymi zmianami w badaniach obrazowych i niekiedy nieprawidłowymi wynikami testów oceniających wydolność narządu (25). Obraz kliniczny choroby oraz zmiany histopatologiczne w narządzie nie pozwalają na rozróżnienie jej czynnika etiologicznego (26). Historia naturalna dziedzicznego zapalenia trzustki oraz związek genotypu z fenotypem opisywane były w kilku pracach opartych na dużych grupach chorych i całych rodzin dotkniętych mutacją w genie PRSS1 (1, 2, 16, 23, 27).
W pracy Howesa i wsp. przeanalizowano dane pacjentów z rejestru EUROPAC, który objął 418 osób ze 112 rodzin, pochodzących z 14 krajów europejskich (16). W tym miejscu podkreślić należy znaczenie podobnych krajowych czy międzynarodowych rejestrów, które poprzez gromadzenie danych o tej rzadkiej chorobie pozwalają znaleźć odpowiedzi na pytania o przebieg choroby, relację genotypu z manifestacją kliniczną czy częstość występowania największego zagrożenia w HP – raka trzustki.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Rebours V, Bourton-Ruault M, Schnee M et al.: The natural history of hereditary pancreatitis: a national series. Gut 2009; 58: 97-103.
2. Joergensen MT, Brusgaard K, Cruger DG et al.: Genetic, epidemiological and clinical aspects of hereditary pancreatitis: a population-based cohort study in Denmark. Am J Gastroenterol 2010; 105: 1876-1883.
3. Comfort MW, Steinberg AG: Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 1952; 21: 54-63.
4. Whitcomb DC, Preston RA, Aston CE et al.: A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology 1996; 110: 1975-1980.
5. Le Bodic L, Schnee M, Georgelin T et al.: An exceptional genealogy for hereditary chronic pancreatitis. Dig Dis Sci 1996; 41: 1504-1510.
6. de las Heras-Castano G, Castro-Senosiain B, Fontalba A et al.: Hereditary pancreatitis: clinical features and inheritance characteristics of the R122C mutation in the cationic trypsinogen gene (PRSS1) in six Spanish families. JOP 2009; 10: 249-255.
7. Whitcomb DC, Gorry MC, Preston RA et al.: Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996; 14: 141-145.
8. Gorry MC, Gabbaizedeh D, Furey W et al.: Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology 1997; 113: 1063-1068.
9. Teich N, Mossner J, Keim V: Mutations of the cationic trypsinogen in hereditary pancreatitis. Hum Mutat 1998; 12: 39-43.
10. Ferec C, Raguenes O, Salomon R et al.: Mutations in the cationic trypsinogen gene and evidence for genetic heterogeneity in hereditary pancreatitis. J Med Genet 1999; 36: 228-232.
11. Witt H, Luck W, Becker M: A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis. Gastroenterology 1999; 117: 7-10.
12. Pfutzer RH, Myers E, Applebaum-Shapiro S et al.: Novel cationic trypsinogen (PRSS1) N29T and R122C mutations cause autosomal dominant hereditary pancreatitis. Gut 2002; 50: 271-272.
13. Simon P, Weiss FU, Sahin-Toth M et al.: Hereditary pancreatitis caused by a novel PRSS1 mutation (Arg-122→Cys) that alters autoactivation and autodegradation of cationic trypsinogen. J Biol Chem 2002; 277: 5404-5410.
14. Oracz G: Przewlekłe zapalenie trzustki u dzieci – diagnostyka i leczenie. Instytut „Pomnik-Centrum Zdrowia Dziecka”, Warszawa 2012.
15. Braganza J, Lee S, MCloy R, McMahon M: Chronic Pancreatitis. Lancet 2011; 377: 1184-1197.
16. Howes N, Lerch M, Greenhalf W et al.: Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004; 2: 252-261.
17. Witt H, Apte MV, Keim V, Wilson JS: Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis and therapy. Gastroenterology 2007; 132: 1557-1573.
18. Ooi CY, Gonska T, Durie PR, Freedman SD: Genetic testing in pancreatitis. Gastroenterology 2010; 138: 2202-2206.
19. Rosendahl J, Witt H, Szmola R et al.: Chymotrypsin C (CTRC) alterations that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet 2008; 40: 78-82.
20. Witt H, Beer S, Rosendahl J et al.: Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet 2013; 45: 1216-1220.
21. Sahin-Toth M, Toth M: Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 2000; 278: 286-289.
22. Szabo A, Sahin-Toth M: Increased Activation of Hereditary Pancreatitis- Human Cationic Tryspinogen Mutants in Presence of Chymotrypsin C. J Biol Chem 2012; 287: 20701-20710.
23. Lowenfels AB, Maisonneuve P, Dimango EP et al.: Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997; 89: 442-446.
24. Zuk K, Czkwianianc E, Degowska M et al.: Zalecenia diagnostyczne i terapeutyczne w przewlekłym zapaleniu trzustki. Rekomendacje Grupy Roboczej Konsultanta Krajowego w dziedzinie Gastroenterologii i Polskiego Klubu Trzustkowego. Przegl Gastroenterol 2011; 6: 339-352.
25. Yadav D, Whitcomb DC: The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol 2010; 7: 131-145.
26. Shrikhande SV, Martignoni ME, Shrikhande M et al.: Comparison of histological features and inflammatory cell reaction in alcoholic, idiopatic and tropical chronic pancreatitis. Br J Surg 2003; 90: 1565-1572.
27. Keim V, Bauer N, Teich N et al.: Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am J Med 2001; 111: 622-626.
28. Ceppa EP, Pitt HA, Hunter JL et al.: Hereditary pancreatitis: endoscopic and surgical management. J Gastrointest Surg 2013; 17: 847-857.
29. Lowenfels AB, Maisonneuve P, Cavallini G et al.: Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Endg J Med 1993; 328: 1433-1437.
30. Malka D, Hammel P, Maire F et al.: Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002; 51: 849-852.
31. Rebours V, Bourton-Ruault M, Schnee M et al.: Risk of pancreatic carcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol 2008; 103: 111-119.
32. Rebours V, Bourton-Ruault M, Jooste V et al.: Mortality rate and risk factors in patients with hereditary pancreatitis: uni- and multidimensional analyses. Am J Gastroenterol 2009; 104: 2312-2317.
33. Urlich CD: Consensus Committees of the European Registry of Hereditary Pancreatic Diseases, Midwest Multi-Center Pancreatic Study Group, International Association of Pancreatology. Pancreatic Cancer in hereditary pancreatitis: consensus guidelines for prevention, screening and treatment. Pancreatology 2001; 1: 416-422.