Karolina Raczkowska-Łabuda, *Lidia Zawadzka-Głos
Primary ciliary dyskinesia: how to diagnose, how to treat
Department of Pediatric Otolaryngology, Medical University of Warsaw, Poland
Head of Department: Lidia Zawadzka-Głos, MD, PhD
Summary
Characteristics of the syndrome. Underlining the diagnostic and therapeutic difficulties. PCD symptoms include ARDS at the age of early – infancy, recurrent lower respiratory tract infections, chronic rhinosinusitis and otitis media, or impaired fertility. PCD requires differentiation with atypical asthma, bronchiectasis and cystic fibrosis. Diagnostic algorithm consist of cascade of tests (brush cytology/bronchoscopic samples, cilia motility evaluation, function and structure assesment with electron microscopy, immunochemical testing, genetic testing) preceded by screening tests (saccharin, measurement nNO). 1. The primary ciliary dyskinesia is rarely taking under consideration in the differential diagnosis of chronic/recurrent upper respiratory tract infections. 2. Available screening tests do not include target group of patients (< 12 y.o.). 3. No recommendations for the type and methods of obtaining material for testing and methods of its transportation. 4. The basic diagnostic limitation is high cost of a conclusive tests. 5. There is necessity to differentiate primary and secondary ciliary dyskinesia. 6. No general algorithm running patients diagnosed with PCD – the mandatory introduction of standard therapy.
INTRODUCTION
Primary ciliary dyskinesia (PCD), also known as immotile cilia syndrome is a rare, genetically determined disease (predominantly inherited as an autosomal recessive) mostly interfering with upper and lower respiratory tract. It is associated in 40-50% with visceral situs inversus and other forms of heterotaxy (1). Symptoms of the disease directly result from impaired ciliary clearance.
Unfortunately disorders due to dysmotile cilia have been poorly studied in children. Therapeutic strategies are taken from the algorithms developed for the treatment of cystic fibrosis (CF). Lower requirement for more PCD research is the consequence.
DISCUSSION
CD is determined by a number of different genetic ciliary defects (tab. 1) that result in ineffective mucociliary clearance.
Table 1. Genes involved in primary ciliary dyskinesia (PCD). Based on (8).
| Gene | Locus | Defective structure | Phenotype |
| NAH5 | 5p15 | ODA | PCD + KS |
| DNAI1 | 9p21- p13 | ODA | PCD + KS |
| DNAH11 | 7p15.3- 21 | Normal | PCD + KS |
| TXNDC3 | 7p14.1 | ODA | KS |
| DNAI2 | 17q25.1 | ODA | PCD + KS |
| KTU | 14q21.3 | ODA+IDA | PCD + KS |
| RPGR | Xp21.1 | Variable | PCD with retinitis pigmentosa |
| OFD1 | Xp22 | Not known | PCD with mental retardation |
| RSPH9 | 6p21 | CP | PCD |
| RSPH4A | 6q22 | CP | PCD |
ODA – outer dynein arm, IDA – inner dynein arm, CP – central pair, KS – Kartagener’s syndrome
Most patients with PCD have symptoms since their birth or early infancy (2), but the diagnosis is usually postponed (3). Significant number of patients are never diagnosed (4). It seems likely (but stays unproven) that early diagnosis of PCD is important as deterioration in lung function can largely be prevented by specialist respiratory care (5).
How to recognise a young child at risk of developing bronchiectasis? The important sign is that of a persistent “wet” sounding cough. Especially if such a cough persists for more than 8 weeks or symptoms return when antibiotic treatment is stopped. In patients with PCD the cough never goes completely away even with treatment. Likewise, chronic upper airway symptoms, such as constant rhinitis (nasal discharge, episodic facial pain and anosmia). Ear symptoms (recurrent otitis media and glue ear) are a frequent complication (85% of patients) that can require multiple interventions, including repeated courses of antibiotics. Testing for PCD should be considered if standard first line investigations to exclude cystic fibrosis (CF) and screening for immunological defects are negative. A number of patients will have a history of unexplained neonatal respiratory distress. Hearing problems are only seen in half of cases. The clinical aspects of PCD, according to patient’s age, are presented in table 2 (3, 6-8).
Table 2. PCD symptoms displayed by various age groups. Based on (8).
| | Symptoms |
| Antenatal | Situs inversustotalis or heterotaxy on antenatal ultrasoundscanning;
25% of individuals with situs inversustotalis have PCD.
The prevalence of PCD within the heterotaxic subclass is unknown |
| 40-50% of PCD patients present with situs inversustotalis (Kartagener’s syndrome in PCD) |
| 6% show heterotaxy (situs ambiguus) |
| Mild fetal cerebral ventriculomegaly |
| Neonatal | > 75% of full-term neonates with PCD exhibit neonatal respiratory distress requiring supplemental oxygen for days to weeks |
| Continuous rhinorrhoea from the first day of life |
| Mirror-image organ arrangement and other forms of heterotaxy |
| Hydrocephalus may occur in some individuals with PCD, and may reflect dysfunctional ependymal cilia |
| Childhood | Chronic productive or wet-sounding cough, associated or not with recurrent atelectasis or pneumonia |
| Atypical asthma that is nonresponsive to treatment, especially if a wet-sounding cough is present |
| Idiopathic bronchiectasis |
| Daily rhinitis without remission; nasal polyps are rare at this age |
Severe chronic sinusitis in older children,
Otitis media with effusion,
Hearing loss |
| Adolescence and adult life | Same as for childhood |
| Bronchiectasis more evident in adulthood (83%) |
| Chronic mucopurulent sputum production is common |
| Digital clubbing may also be found |
| Pulmonary function tests usually show a progressive obstructive or mixed pattern |
| Nasal polyposis and halitosis |
| Infertility in males (-50%) due to immotility of spermatozoa ectopic pregnancy and subfertility in females |
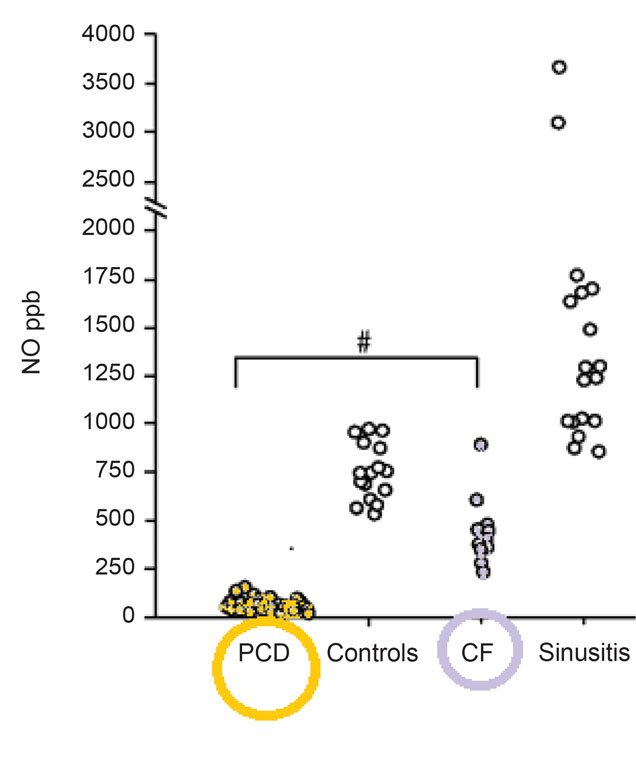
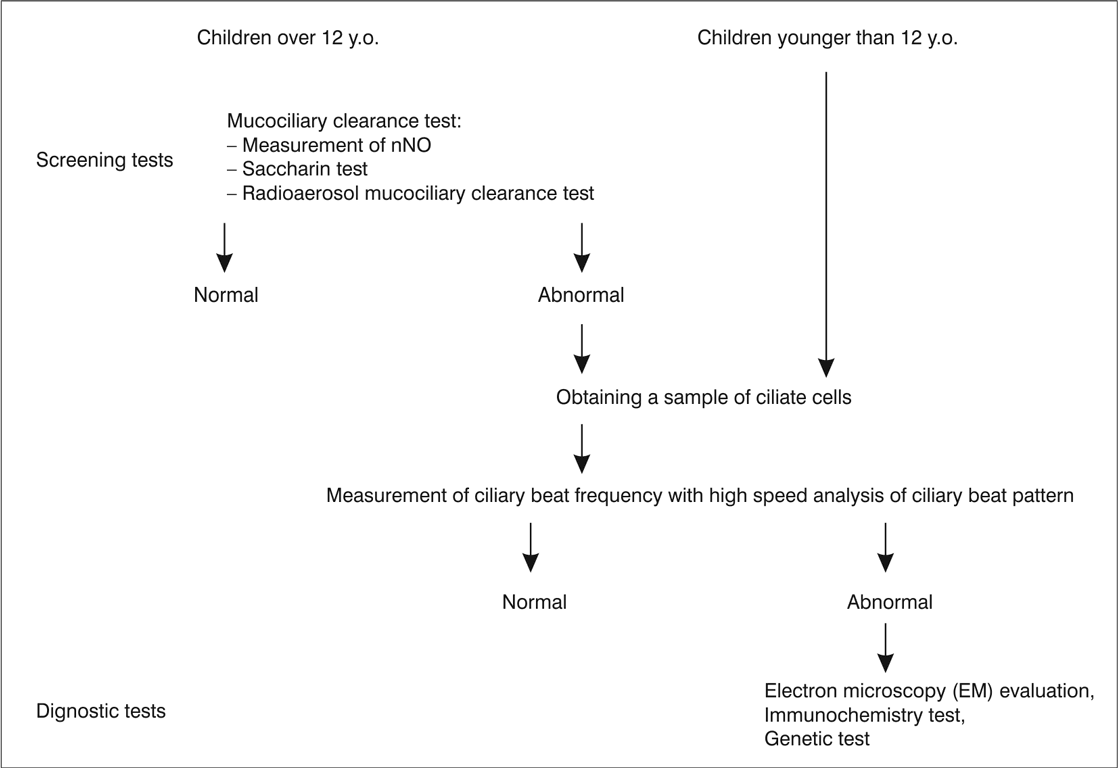
Diagnosis, due to the lack of standardisation and inaccessibility stays challenging either for the patient or the paediatrician. Screening tests for PCD do exist but they are uncorrelated. There are number of problems and the biggest is that most of the screening tests are unreliable in children. For the moment, the most sensitive and specific screening test for PCD is measurement of nasal nitric oxide (fig. 1) (9-11) but even along special paediatric program it is dedicated for children older than 5 years of age (as well as radioaerosol mucociliary clearance tests). What specialist centres may offer to their patients as diagnostic test? First: obtaining a sample of ciliate cells by nasal brushing or bronchoscopic samples. Then analysis of ciliary function by ciliary beat pattern and frequency analysis, evaluation of the cilia’s structure on electron microscopy and even genetic analysis (12). Diagnostic algorithm is presented on figure 2.
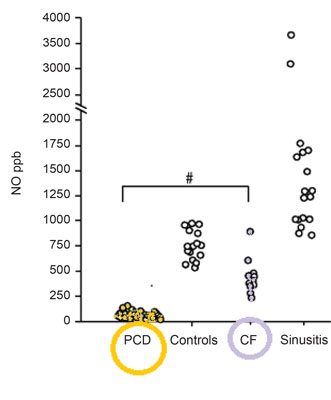
Fig. 1. Nasal nitric oxidemeasurement due to CF and sinusitis test results. Based on (10).
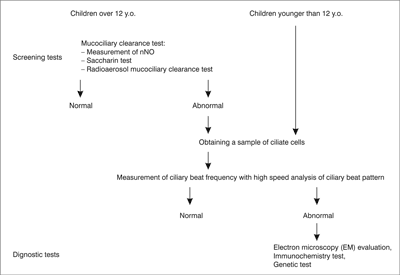
Fig. 2. PCD diagnostic algorithm; a proposition.
Restore or maintain normal lung function is the main aim of therapy for PCD. Unfortunately there are no randomised trials of adequate treatment. Therefore recommendations are based on a very low level evidence. The clinician should abort therapy that is not working. Main, valid guidelines for the therapy are:
– airway clearance by combinations of physiotherapy and physical exercise should be encouraged and regularly prescribed in children with PCD (11, 12),
– high-dose oral antibiotics should be prescribed at the first sign of worsening respiratory symptoms or deterioration in lung function; if persistent respiratory symptoms do not respond to oral antibiotics, then intravenous therapy should be given (8),
– anti-inflammatory strategies, such as alternate-day prednisolone or inhaled corticosteroids, should not be used (12, 13),
– Otitis media with effusion should be managed conservatively, with regular audiological assessment, hearing aids and hearing therapy (14, 15),
– the use of ventilation tubes (grommets) should be avoided where possible (8, 14, 15),
– regular (≥ 3 monthly) sputum or cough-swab cultures should be performed based on clinic and hospital or local colonisation (8).
To deliver best care, patients with PCD should be seen for either full or shared care in a centre specialising in the condition. There is urgent need to publicize information about syndrome itself, about reference centres and diagnostic methods. Further studies are clearly needed to establish general consensus statement.
CONCLUSIONS
1. The primary ciliary dyskinesia is rarely taking under consideration in the differential diagnosis of chronic/recurrent upper respiratory tract infections.
2. Available screening tests do not include target group of patients (< 12 y.o.).
3. No recommendations for the type and methods of obtaining material for testing and methods of its transportation.
4. The basic diagnostic limitation is high cost of a conclusive tests.
5. There is necessity to differentiate primary and secondary ciliary dyskinesia.
6. No general algorithm running patients diagnosed with PCD – the mandatory introduction of standard therapy.
Piśmiennictwo
1. O’Callaghan C, Chilvers M, Hogg C et al.: Diagnosing primary ciliary dyskinesia. Thorax 2007; 62: 656-657. 2. Greenstone M, Rutman A, Dewar A et al.: Primary ciliary dyskinesia: cytological and clinical features. Q J Med 1988; 67: 405-423. 3. Noone PG, Leigh MW, Sannuti A et al.: Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med 2004; 169: 459-467. 4. Bush A, Cole P, Hariri M et al.: Primary ciliary dyskinesia: diagnosis and standards of care. Eur Respir J 1998; 12: 982-988. 5. Ellerman A, Bisgaard H: Longitudinal study of lung function in a cohort of primary ciliary dyskinesia. Eur Respir J 1997; 10: 2376-2379. 6. Coren ME, Meeks M, Morrison I et al.: Primary ciliary dyskinesia: age at diagnosis and symptom history. Acta Paediatr 2002; 91: 667-669. 7. Ferkol T, Leigh M: Primary ciliary dyskinesia and newborn respiratory distress. Semin Perinatol 2006; 30: 335-340. 8. Barbato A, Frischer T, Kuehni CE et al.: Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. ERJ 2009 Dec 1; 34(6): 1264-1276. 9. Marthin JK, Nielsen KG: Choice of nasal nitric oxide technique as first-line test for primary ciliary dyskinesia. ERJ 2011 Mar 1; 37(3): 559-565. 10. Wodehouse T, Kharitonov SA, Mackay IS et al.: Nasal nitric oxide measurements for the screening of primary ciliary dyskinesia. ERJ 2003 Jan 1; 21(1): 43-47. 11. Corbelli R, Bringolf-Isler B, Amacher A et al.: Nasal nitric oxide measurements to screen children for primary ciliary dyskinesia. Chest 2004; 126(4): 1054-1059. 12. Phillips GE, Thomas S, Heather S et al.: Airway response of children with primary ciliary dyskinesia to exercise and β2-agonist challenge. Eur Respir J 1998; 11: 1389-1391. 13. Balfour-Lynn IM, Lees B, Hall P et al.: Multicenter randomized controlled trial of withdrawal of inhaled corticosteroids in cystic fibrosis. Am J Respir Crit Care Med 2006; 173: 1356-1362. 14. Majithia A, Fong J, Hariri M et al.: Hearing outcomes in children with primary ciliary dyskinesia – a longitudinal study. Int J Pediatr Otorhinolaryngol 2005; 69: 1061-1064. 15. Hadfield PJ, Rowe-Jones JM, Bush A et al.: Treatment of otitis media with effusion in children with primary ciliary dyskinesia. Clin Otolaryngol Allied Sci 1997; 22: 302-306.