© Borgis - Postępy Nauk Medycznych 2/2009, s. 149-156
Jakub Karczmarski, Michał Mikula, Ewa Hennig, Tymon Rubel, *Jerzy Ostrowski
Perspektywy poszukiwania biomarkerów chorobowych w peptydomie osocza lub surowicy krwi z użyciem spektrometrii mas
Perspectives of mass spectrometry-based disease biomarkers discovery in plasma or serum peptidome
Centrum Medyczne Kształcenia Podyplomowego, Klinika Gastroenterologii i Hepatologii w Centrum Onkologii – Instytut im. Marii Skłodowskiej-Curie
Kierownik Kliniki: prof. dr hab. med. Jarosław Reguła
Streszczenie
Dzięki ogromnemu postępowi, jaki dokonuje się w technikach proteomicznych, a w szczególności w spektrometrii mas, proteomika jest powszechnie uważana za niezwykle obiecującą technologię w badaniach biomedycznych. Jednym z jej głównych zastosowań stało się poszukiwanie biologicznych markerów chorobowych. Klinicznie przydatny marker powinien być łatwo mierzalny w tkance lub płynie ustrojowym, których pobranie nie powinno być uciążliwe dla chorego, a tym samym badanie diagnostyczne lub predykcyjne powinno być w pełni przez niego akceptowane. Badanie niskocząsteczkowego proteomu osocza i surowicy zyskuje coraz większe zainteresowanie, gdyż uważa się, że to właśnie w tym obszarze nazwanym peptydomem, mogą znajdować się nowe, jeszcze nie odkryte biomarkery. Celem tego artykułu jest przegląd najnowszych metod i trendów w badaniu proteomu/peptydomu osocza i surowicy oraz przedstawienie możliwości i ograniczeń zastosowania spektrometrii mas do poszukiwania peptydowych biomarkerów chorobowych, w szczególności w onkologii.
Summary
Recent immense progress in proteomic technologies, especially in mass spectrometry, makes proteomics a promising field for biomedical research. One of rapidly developing branches in proteomics is the discovery of biological disease markers. Clinically useful biomarkers should be easily detectable in tissues or in bodily fluids that are accessible for sampling without subsequent inconvenience to a patient. The low molecular weight fraction of plasma and serum, called peptidome, is gaining a growing interest, as it is considered to be the source of currently undiscovered biomarkers. This article intends to review the newest methods and trends in plasma and serum peptidome research and to show capabilities and limitations as well as current applications of mass spectrometry in peptide disease biomarker discovery, taking a special focus on oncology.

WPROWADZENIE
Termin proteomika po raz pierwszy został użyty w 1994 roku i w analogii do genomiki, oznaczał zakrojone na szeroką skalę badania mające na celu poznanie ogółu białek komórkowych (1). Dzisiaj proteomika zajmuje się również określaniem struktury, izoform oraz modyfikacji potranslacyjnych białek, badaniem mechanizmów aktywności, funkcji i usytuowania białek w szlakach metabolicznych komórki, a także analizą interakcji z innymi białkami, struktury tworzonych kompleksów oraz ich lokalizacji śródkomórkowej.
Jedną z podstawowych technik stosowanych w proteomice jest spektrometria mas (MS), ponieważ pozwala ona na pomiar masy cząsteczek w stężeniach attomolowych, do niedawna nieosiągalnych w analizie. Spektrometr mas składa się z trzech podstawowych części: źródła jonów, analizatora rozdzielającego jony pod względem stosunków ich masy do ładunku oraz detektora zliczającego liczbę jonów danego rodzaju. Bardzo szybki rozwój tej techniki został zapoczątkowany w latach 90-tych, dzięki wprowadzeniu nowych technik jonizacji, takich jak:
– jonizacja przez desorpcję laserową w matrycy (MALDI, ang. Matrix Assisted Laser Desorption Ionisation), w której stosuje się jonizację wiązką laserową o tak dobranej energii, aby nie doprowadzać do fragmentacji cząsteczek (tzw. metoda łagodnej jonizacji), a jedynie do ich „wybijania” ze specjalnie przygotowanej matrycy. Matryca absorbuje energię lasera, która następnie jest przekazywana do analizowanych cząsteczek (2),
– elektrorozpylanie (ESI, ang. Electrospray), polegające na rozpylaniu cieczy, zawierającej badaną substancję, w polu elektrycznym o wysokim napięciu (zwykle 1-5 kV). Jest to również technika łagodnej jonizacji – zwykle nie powoduje fragmentacji badanych cząsteczek (2).
Za pionierskie prace nad ESI i MALDI, John Fenn i Koichi Tanaki zostali uhonorowani w 2002 roku nagrodą Nobla w dziedzinie chemii (3). Dalsze zwiększenie czułości analiz w MS osiągnięto dzięki sprzężeniu ESI, a od niedawna również MALDI, z chromatografią cieczową (4).
Dzięki ogromnemu postępowi, jaki dokonuje się w technikach proteomicznych, a w szczególności w MS, proteomika jest powszechnie uważana za niezwykle obiecującą technologię w badaniach biomedycznych. Jednym z jej głównych zastosowań stało się poszukiwanie biologicznych markerów chorobowych. Szczególnie interesującym źródłem biomarkerów mogą być płyny ustrojowe. Będąc w stałym i bliskim kontakcie z tkankami, płyny ustrojowe stają się rezerwuarem polipeptydów wydzielanych przez tkanki, a ich skład zależy od metabolicznego stanu organizmu. Dodatkowo, pobieranie płynów ustrojowych cechuje niska inwazyjność i niewielkie koszty oraz łatwość przechowywania próbek i ich przetwarzania. Jednakże odkrycie nowych biomarkerów chorobowych wymaga zrozumienia specyfiki płynów ustrojowych oraz ścisłej standaryzacji wielu etapów analizy.
MOŻLIWOŚCI I OGRANICZENIA SPEKTROMETRII MAS W BADANIACH BIOMEDYCZNYCH
Osocze i surowica krwi jako potencjalne źródło biomarkerów
Liczba i skład polipeptydów w krwioobiegu zmienia się dynamicznie w zależności od stanu organizmu, odzwierciedlając zachodzące w nim zmiany, tak fizjologiczne, jak i chorobowe (5). Docierając do wszystkich regionów ciała, krew zbiera z tkanek wydzielane komórkowe białka i metabolity, stając się potencjalnie ważnym źródłem biomarkerów chorobowych.
Dla poszukiwania markerów białkowych we krwi ważny jest wybór czy źródłem białek będzie osocze czy też surowica, bowiem ich składy białkowe w dużym stopniu różnią się od siebie (6). Różnica ta wynika przede wszystkim z usunięcia z surowicy znacznych ilości fibrynogenu oraz białek specyficznie bądź niespecyficznie związanych z procesem wykrzepiania krwi.
Grupy badawcze skupione w międzynarodowej organizacji HUPO (ang. Human Proteome Organisation), na podstawie wyników otrzymanych w pilotażowej fazie projektu Proteom Osocza (ang. Plasma Proteome Project), rekomendują używanie do badań osocza zamiast surowicy krwi, ze względu na niższy poziom degradacji białek zachodzącej ex vivo, a tym samym większą stabilność składu białkowego próbek (7). Jednakże coraz częściej pojawiają się również opinie przemawiające za stosowaniem surowicy jako źródła potencjalnych biomarkerów peptydowych, właśnie ze względu na procesy zachodzące ex vivo, które według zwolenników stosowania do badań surowicy wydają się być jednym z mechanizmów generowania biomarkerów (8). Tak więc, oprócz wątpliwości: co i jak pobierać, jak transportować, jak przechowywać i jak wstępnie przygotowywać próbki do analizy, rodzi się kolejne pytanie: badać proteom czy peptydom, osocza czy surowicy?
Standaryzacja procedur przygotowania prób do analiz
W badaniach proteomów osocza i surowicy krwi w poszukiwaniu biomarkerów niezmiernie ważna jest powtarzalność eksperymentów i możliwość odtworzenia wyników analiz w innych ośrodkach badawczych. Badania własne i innych autorów wskazują, że czas i temperatura wykrzepiania krwi, wielokrotność rozmrażania i zamrażania próbek surowicy, a także rodzaj i ilość stosowanych inhibitorów proteaz (niektóre z nich mogą interferować z analizą w spektrometrze mas) mają znaczący wpływ na uzyskiwane wyniki analiz (9, 10). Cel, jaki stawia sobie HUPO, upowszechniając swoje wyniki i obserwacje, to m.in. opracowanie standardowych protokołów i metod, które umożliwią porównywanie wyników otrzymanych w różnych laboratoriach.
Dynamiczny zakres stężeń białek krwi
Proteomy surowicy i osocza krwi charakteryzuje złożony skład oraz dynamiczny zakres stężeń występujących w nich białek. Ich pełną analizę proteomiczną istotnie utrudnia obecność kilku białek, które występują w bardzo wysokich stężeniach. Do tych białek zaliczamy m.in. albuminę, immunoglobuliny, alfa-1-antytrypsynę, fibrynogen, transferynę oraz haptoglobinę. Szacuje się, że zaledwie 22 białka stanowią niemal 99% masy białkowej osocza i surowicy (ryc. 1). Sama albumina stanowi ponad 50% masy białkowej, a poziom jej stężenia w surowicy w porównaniu z białkami sygnałowymi, występującymi w ilościach śladowych, przekracza 10 rzędów wielkości (5). Podczas rutynowej analizy proteomicznej, peptydy uzyskiwane z białek najczęściej występujących całkowicie maskują obecność peptydów pochodzących z białek o niższym stężeniu, wśród których spodziewamy się znaleźć biomarkery. Dlatego też poszukiwanie potencjalnych biomarkerów peptydowych wymaga usunięcia z analizowanej mieszaniny białek występujących w niej w dużych stężeniach, np. poprzez stosowanie kolumn wypełnionych złożem z przeciwciałami skierowanymi przeciwko najczęściej występującym białkom krwi (6).
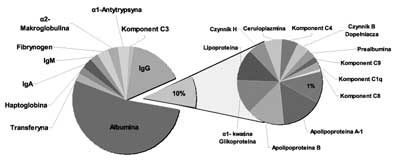
Ryc. 1. Udział najczęściej występujących białek krwi, wg Tirumalai i wsp. (11).
Metody poszukiwania biomarkerów z użyciem spektrometrii mas
Prosty eksperyment z użyciem tandemowego spektrometru mas sprzężonego z wysokosprawną chromatografią cieczową (LC-MS/MS) zazwyczaj generuje listę kilkuset białek. Dla pełniejszej identyfikacji proteomu stosuje się złożone, wieloetapowe metody proteomiczne, które najczęściej obejmują wstępne frakcjonowanie białek, wielowymiarowy rozdział polipeptydów i ich ostateczną identyfikację w MS. W tabeli 1 przedstawiono przykłady różnych połączeń technik proteomicznych stosowanych w badaniach proteomów surowicy i osocza wraz z liczbą zidentyfikowanych w nich białek.
Tabela 1. Przykłady zastosowania różnych technik proteomicznych do badania białek krwi.
| Badany proteom | Zastosowana metodologia | Liczba zidentyfikowanych białek | Piśmiennictwo |
| surowica | SCX*/LC-IT MS | 490 | (49) |
| osocze | Nano-LC/IT MS | 800-1682 | (50) |
| surowica | wstępne frakcjonowanie SEC, AEC LC-ESI-MS/MS | 742 | (51) |
| osocze/surowica | wstępne frakcjonowanie IEF,1DE RPLC-MS/MS | 575 (osocze)
2890 (surowica) | (52) |
| surowica | IEF/LC-MS/MS | 437 | (13) |
| surowica | wstępne frakcjonowanie IEF-SCX LC-MS/MS | 1444 | (14) |
| surowica | LC-FT-ICR MS | 722 | (53) |
| osocze | wstępne frakcjonowanie SCX RPLC-MS/MS | 804 | (54) |
| osocze/surowica | LAC/LC-MS/MS | 150 (białka glikozylowane) | (20) |
| osocze | SCX-RPLC-MS/MS | 303 (białka glikozylowane) | (22) |
*SCX – chromatografia kationowymienna, LC – chromatografia cieczowa, IT – pułapka jonowa, MS – spektrometria mas, SEC – chromatografia wykluczania, AEC – chromatografia anionowymienna, ESI – elektrorozpylanie, IEF – izoelektroogniskowanie, 1DE – elektroforeza w denaturującym żelu poliakryloamidowym, RPLC – chromatografia cieczowa w odwróconej fazie, LAC – chromatografia powinowactwa z użyciem lektyn, FT-ICR – analizator cyklotronowego rezonansu jonów z furierowską transformacją wyników.
Do najczęściej stosowanych metod frakcjonowania białek osocza i surowicy należą: chromatografia cieczowa w odwróconej fazie (RPLC), chromatografia jonowymienna (kationowymienna SCX i anionowymienna AEC), chromatografia wykluczania (SEC), elektroforeza w denaturującym żelu poliakryloamidowym (SDS-PAGE), elektroforeza kapilarna (EC) i izoelektroogniskowanie (IEF) (6). Łączenie ze sobą różnych technik frakcjonowania może dodatkowo zwiększać ich rozdzielczość, pozwalając na wielowymiarowe frakcjonowanie próbek i dalsze upraszczanie ich składu białkowego przed analizą MS.
W projekcie HUPO/PPP przeprowadzono szereg porównań w celu uzyskania najbardziej wydajnej metody frakcjonowania białek, poprzedzającej analizę MS. Okazało się, że na identyfikację największej liczby białek surowicy pozwalają metody sprzężone z SCX oraz z IEF, ponadto każda z tych metod identyfikuje z osobna białka dla siebie unikatowe (12). W eksperymencie, w którym połączono IEF z LC-MS/MS zidentyfikowano w surowicy 844 peptydy, które odpowiadały 437 białkom (13), natomiast połączenie IEF z SCX i LC-MS/MS pozwoliło na rozpoznane aż 1444 białek (14). We wstępnym podsumowaniu projektu PPP, HUPO przedstawiła listę 9504 białek zidentyfikowanych na podstawie co najmniej jednego peptydu, w tym 3020 białek identyfikowanych na podstawie co najmniej dwóch peptydów (7).
Niskocząsteczkowy proteom krwi
Badanie niskocząsteczkowego proteomu osocza i surowicy zyskuje coraz większe zainteresowanie, gdyż uważa się, że to właśnie w tym obszarze proteomu, nazwanym peptydomem, mogą znajdować się nowe, jeszcze nie odkryte biomarkery. Za „niskocząsteczkową frakcję” proteomu krwi przyjęto uważać peptydy i białka o masie poniżej 30 kDa (15). W jej skład wchodzi szereg ważnych fizjologicznie białek, takich jak: cytokiny, chemokiny, białkowe hormony i czynniki wzrostu. Peptydy występujące w krwioobiegu mogą także powstawać jako produkty degradacji białek, w wyniku aktywności endogennych proteaz (16). Frakcję niskocząsteczkową proteomu krwi można otrzymać przez usunięcie polipeptydów o masie powyżej 30 kDa, stosując kolumny filtracyjne o takim punkcie odcięcia. W celu uwolnienia polipeptydów związanych przez wysokocząsteczkowe białka nośnikowe, filtrowanie przeprowadza się w warunkach denaturujących (15).
Wiele składników niskocząsteczkowego proteomu krwi jest związanych z białkami nośnikowymi, do których zaliczamy przede wszystkim albuminę. Uważa się, że albumina pełni rolę „gąbki molekularnej” dla peptydów i niskocząsteczkowych białek, dzięki czemu nie są one usuwane przez układ filtracyjny nerek i dłużej pozostają w krwioobiegu (17). Dla przykładu, w osoczu chorych na raka jajnika wyróżniono ponad 800 peptydów wiązanych przez albuminę, w tym fragmenty białka komórkowego BRCA2 (18). Wydaje się więc, że frakcja niskocząsteczkowych białek i peptydów wiązanych przez białka nośnikowe, może również stanowić ważne źródło potencjalnych biomarkerów chorobowych.
Białka glikozylowane
Izolowanie białek podlegających specyficznym modyfikacjom potranslacyjnym łańcuchów bocznych aminokwasów, stanowi jedno z możliwych podejść do badania białek obecnych we krwi w niskich stężeniach. Modyfikacje te odgrywają zasadniczą rolę w regulacji wielu podstawowych procesów biologicznych, a zaburzenie ich normalnego przebiegu ma wpływ na powstawanie wielu chorób, w tym również nowotworowych. Glikozylacja jest jedną z najczęściej występujących modyfikacji białek, a zmiany w składzie podstawianych reszt wielocukrowych wydają się być specyficzne dla różnych stanów chorobowych (19).
Znakomita większość białek wydzielanych przez komórki podlega glikozylacji, włączając w to znane już markery nowotworowe, jak PSA, CA125, CA19-9, CA15-3 czy CEA (19). Zidentyfikowanie glikozylowanych peptydów często wymaga jednak ich wzbogacenia przed analizą w spektrometrze mas. Stosuje się w tym celu chromatografię powinowactwa z użyciem lektyn (przy pomocy tej techniki zidentyfikowano ponad 150 glikozylowanych białek w osoczu i surowicy (20)), jak również chromatografię wykluczania (21). Możliwe jest także wcześniejsze enzymatyczne odłączenie reszt wielocukrowych od szkieletu polipeptydowego, przy pomocy glikozydazy F. Analiza otrzymanych w ten sposób reszt wielocukrowych przy użyciu elektroforezy dwukierunkowej (2D-E) i LC-MS/MS pozwoliła na zidentyfikowanie 2053 N-glikopeptydów, reprezentujących 303 glikoproteiny (22). Wykazano, że specyficzny skład wielocukrów obecnych w surowicy może charakteryzować chorych na raka gruczołu krokowego (23) i trzustki (24).
PROTEOMIKA W BADANIACH BIOMEDYCZNYCH
Zastosowanie spektrometrii mas do poszukiwania biomarkerów chorobowych
Wprowadzenie technologii proteomicznych i ciągłe ich udoskonalanie niesie nadzieję na ich zastosowanie w diagnostyce chorób. O rosnącym zainteresowaniu i zapotrzebowaniu na tego typu badania świadczy lawinowy wzrost liczby doniesień i publikacji na ten temat, ukazujących się w światowym piśmiennictwie w ostatnich latach.
Wyniki wielu badań wskazują na istnienie korelacji pomiędzy stężeniem pewnych białek lub peptydów we krwi, a stanem chorobowym badanych osób. Obiecujące wydają się wyniki analiz proteomu surowicy pacjentów cierpiących na choroby autoimmunologiczne (25, 26, 27). Wykazano, że u chorych z przewlekłym wirusowym zapaleniem wątroby (WZW) typu B, określone profile proteomiczne krwi (skład białkowy) korelują ze stopniem zwłóknienia wątroby oraz umożliwiają różnicowanie między zwłóknieniem a marskością wątroby (28). Wśród białek, których zmienność koreluje ze stopniem morfologicznego uszkodzenia wątroby w przebiegu WZW typu B, znajdowano apolipoproteiny A-I i A-IV, alfa-1-antytrypsynę, transtyretynę i topoizomerazę II (29). Podobne zależności obserwowano również dla glikozylowanych białek krwi: osoczowego amyloidu P i ceruloplazminy (30). Z kolei, u chorych z rakiem wątrobowo-komórkowym wykazano zmiany glikozylacji białka GP73 (31).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Wilkins MR et al.: From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (NY) 1996; 14: 61-5.
2. de Hoffmann E, Charette J, Stroobant V: Spektrometria mas. Warszawa, Wydawnictwa Naukowo-Techniczne 1998; 39-41.
3. Cho A, Normile D: Nobel Prize in Chemistry. Mastering macromolecules. Science 2002; 298: 527-8.
4. Covey TR, Huang EC, Henion JD: Structural characterization of protein tryptic peptides via liquid chromatography/mass spectrometry and collision-induced dissociation of their doubly charged molecular ions. Anal Chem 1991; 63: 1193-200.
5. Anderson NL, Anderson NG: The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002; 1: 845-67.
6. Hu S, Loo JA, Wong DT: Human body fluid proteome analysis. Proteomics 2006; 6: 6326-53.
7. Omenn GS et al.: Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics 2005; 5: 3226-45.
8. Villanueva J et al.: Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest 2006; 116: 271-84.
9. Rai AJ et al.: HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 2005; 5: 3262-77.
10. West-Nielsen M et al.: Sample handling for mass spectrometric proteomic investigations of human sera. Anal Chem 2005; 77: 5114-23.
11. Tirumalai RS et al.: Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics 2003; 2: 1096-103.
12. Barnea E et al.: Evaluation of prefractionation methods as a preparatory step for multidimensional based chromatography of serum proteins. Proteomics 2005; 5: 3367-75.
13. Xiao Z et al.: Direct ampholyte-free liquid-phase isoelectric peptide focusing: application to the human serum proteome. Electrophoresis 2004; 25: 128-33.
14. Chan KC et al.: Analysis of the human serum proteome Clinical Proteomics 2004; 101-225.
15. Drake RR, Cazares L, Semmes OJ: Mining the low molecular weight proteome of blood. Proteomics Clin. Appl. 2007; 1: 758-68.
16. Diamandis EP: Peptidomics for cancer diagnosis: present and future. J Proteome Res 2006; 5: 2079-82.
17. Dennis MS et al.: Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem 2002; 277: 35035-43.
18. Lowenthal MS et al.: Analysis of albumin-associated peptides and proteins from ovarian cancer patients. Clin Chem 2005; 51: 1933-45.
19. Bosques CJ, Raguram S, Sasisekharan R: The sweet side of biomarker discovery. Nat Biotechnol 2006; 24: 1100-1.
20. States DJ et al.: Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat Biotechnol 2006; 24: 333-8.
21. Alvarez-Manilla G et al.: Tools for glycoproteomic analysis: size exclusion chromatography facilitates identification of tryptic glycopeptides with N-linked glycosylation sites. J Proteome Res 2006; 5: 701-8.
22. Liu T et al.: Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res 2005; 4: 2070-80.
23. Kyselova Z et al.: Alterations in the serum glycome due to metastatic prostate cancer. J Proteome Res 2007; 6: 1822-32.
24. Zhao J et al.: N-linked glycosylation profiling of pancreatic cancer serum using capillary liquid phase separation coupled with mass spectrometric analysis. J Proteome Res 2007; 6: 1126-38.
25. Liao H et al.: Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum 2004; 50: 3792-803.
26. Kuhn E et al.: Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics 2004; 4: 1175-86.
27. Avasarala JR, Wall MR, Wolfe GM: A distinctive molecular signature of multiple sclerosis derived from MALDI-TOF/MS and serum proteomic pattern analysis: detection of three biomarkers. J Mol Neurosci 2005; 25: 119-25.
28. Poon TC et al.: Prediction of liver fibrosis and cirrhosis in chronic hepatitis B infection by serum proteomic fingerprinting: a pilot study. Clin Chem 2005; 51: 328-35.
29. He QY et al.: Serum biomarkers of hepatitis B virus infected liver inflammation: a proteomic study. Proteomics 2003; 3: 666-74.
30. Comunale MA et al.: Comparative proteomic analysis of de-N-glycosylated serum from hepatitis B carriers reveals polypeptides that correlate with disease status. Proteomics 2004; 4: 826-38.
31. Block TM et al.: Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc Natl Acad Sci U S A 2005; 102: 779-84.
32. Stanley BA et al.: Heart disease, clinical proteomics and mass spectrometry. Dis Markers 2004; 20: 167-78.
33. Marshall J et al.: Processing of serum proteins underlies the mass spectral fingerprinting of myocardial infarction. J Proteome Res 2003; 2: 361-72.
34. Dayal B, Ertel NH: ProteinChip technology: a new and facile method for the identification and measurement of high-density lipoproteins apoA-I and apoA-II and their glycosylated products in patients with diabetes and cardiovascular disease. J Proteome Res 2002; 1: 375-80.
35. Allard L et al.: ApoC-I and ApoC-III as potential plasmatic markers to distinguish between ischemic and hemorrhagic stroke. Proteomics 2004; 4: 2242-51.
36. Yip TT et al.: Protein chip array profiling analysis in patients with severe acute respiratory syndrome identified serum amyloid a protein as a biomarker potentially useful in monitoring the extent of pneumonia. Clin Chem 2005; 51: 47-55.
37. Kang X et al.: Proteomic fingerprints for potential application to early diagnosis of severe acute respiratory syndrome. Clin Chem 2005; 51: 56-64.
38. Petricoin EF et al.: Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002; 359: 572-7.
39. Liotta LA et al.: General Keynote: Proteomic Patterns in Sera Serve as Biomarkers of Ovarian Cancer. Gynecol Oncology 2003; 88: S25-S8.
40. Petricoin EF et al.: Serum proteomic patterns for detection of prostate cancer. J Natl Cancer Inst 2002; 94: 1576-8.
41. Ransohoff DF: Lessons from controversy: ovarian cancer screening and serum proteomics. J Natl Cancer Inst 2005; 97: 315-9.
42. Diamandis EP, van der Merwe DE: Plasma protein profiling by mass spectrometry for cancer diagnosis: opportunities and limitations. Clin Cancer Res 2005; 11: 963-5.
43. Koomen JM et al.: Direct tandem mass spectrometry reveals limitations in protein profiling experiments for plasma biomarker discovery. J Proteome Res 2005; 4: 972-81.
44. van Hensbergen Y et al.: Soluble aminopeptidase N/CD13 in malignant and nonmalignant effusions and intratumoral fluid. Clin Cancer Res 2002; 8: 3747-54.
45. Zhang Z et al.: Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res 2004; 64: 5882-90.
46. Villanueva J et al.: A sequence-specific exopeptidase activity test (SSEAT) for „functional” biomarker discovery. Mol Cell Proteomics 2008; 7: 509-18.
47. Diamandis EP, Kulasingam V, Sardana G: Letter to the Editor about Differential exoprotease activities confer tumor-specific serum peptidome. JCI eletters 2006; http://www.jci.org/eletters/view/26022#sec2.
48. Pepe MS et al.: Phases of biomarker development for early detection of cancer. J Natl Cancer Inst 2001; 93: 1054-61.
49. Adkins JN et al.: Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol Cell Proteomics 2002; 1: 947-55.
50. Shen Y et al.: Ultra-high-efficiency strong cation exchange LC/RPLC/MS/MS for high dynamic range characterization of the human plasma proteome. Anal Chem 2004; 76: 1134-44.
51. Horn A et al.: Multidimensional proteomics of human serum using parallel chromatography of native constituents and microplate technology. Proteomics 2006; 6: 559-70.
52. Tang HY et al.: A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics 2005; 5: 3329-42.
53. Adkins JN et al.: A proteomic study of the HUPO Plasma Proteome Project´s pilot samples using an accurate mass and time tag strategy. Proteomics 2005; 5: 3454-66.
54. Qian WJ et al.: Comparative proteome analyses of human plasma following in vivo lipopolysaccharide administration using multidimensional separations coupled with tandem mass spectrometry. Proteomics 2005; 5: 572-84.
55. Le L et al.: Identification of serum amyloid A as a biomarker to distinguish prostate cancer patients with bone lesions. Clin Chem 2005; 51: 695-707.
56. Koomen JM et al.: Plasma protein profiling for diagnosis of pancreatic cancer reveals the presence of host response proteins. Clin Cancer Res 2005; 11: 1110-8.
57. Yokoi K et al.: Serum amyloid A as a tumor marker in sera of nude mice with orthotopic human pancreatic cancer and in plasma of patients with pancreatic cancer. Int J Oncol 2005; 27: 1361-9.
58. Howard BA et al.: Identification and validation of a potential lung cancer serum biomarker detected by matrix-assisted laser desorption/ionization-time of flight spectra analysis. Proteomics 2003; 3: 1720-4.
59. Won Y et al.: Pattern analysis of serum proteome distinguishes renal cell carcinoma from other urologic diseases and healthy persons. Proteomics 2003; 3: 2310-6.
60. Chen YD et al.: Artificial neural networks analysis of surface-enhanced laser desorption/ionization mass spectra of serum protein pattern distinguishes colorectal cancer from healthy population. Clin Cancer Res 2004; 10: 8380-5.
61. Poon TC et al.: Comprehensive proteomic profiling identifies serum proteomic signatures for detection of hepatocellular carcinoma and its subtypes. Clin Chem 2003; 49: 752-60.
62. Paradis V et al.: Identification of a new marker of hepatocellular carcinoma by serum protein profiling of patients with chronic liver diseases. Hepatology 2005; 41: 40-7.
63. Li J et al.: Proteomics and bioinformatics approaches for identification of serum biomarkers to detect breast cancer. Clin Chem 2002; 48: 1296-304.
64. Jung SS et al.: The HCCR oncoprotein as a biomarker for human breast cancer. Clin Cancer Res 2005; 11: 7700-8.
65. Semmes OJ et al.: Discrete serum protein signatures discriminate between human retrovirus-associated hematologic and neurologic disease. Leukemia 2005; 19: 1229-38.
66. Ebert MP et al.: Identification of the thrombin light chain a as the single best mass for differentiation of gastric cancer patients from individuals with dyspepsia by proteome analysis. J Proteome Res 2005; 4: 586-90.
